

iTRAQ-Based Proteomic Analysis of APP Transgenic Mouse Urine Exosomes
 , , and
, , and
Abstract
:1. Introduction
2. Results
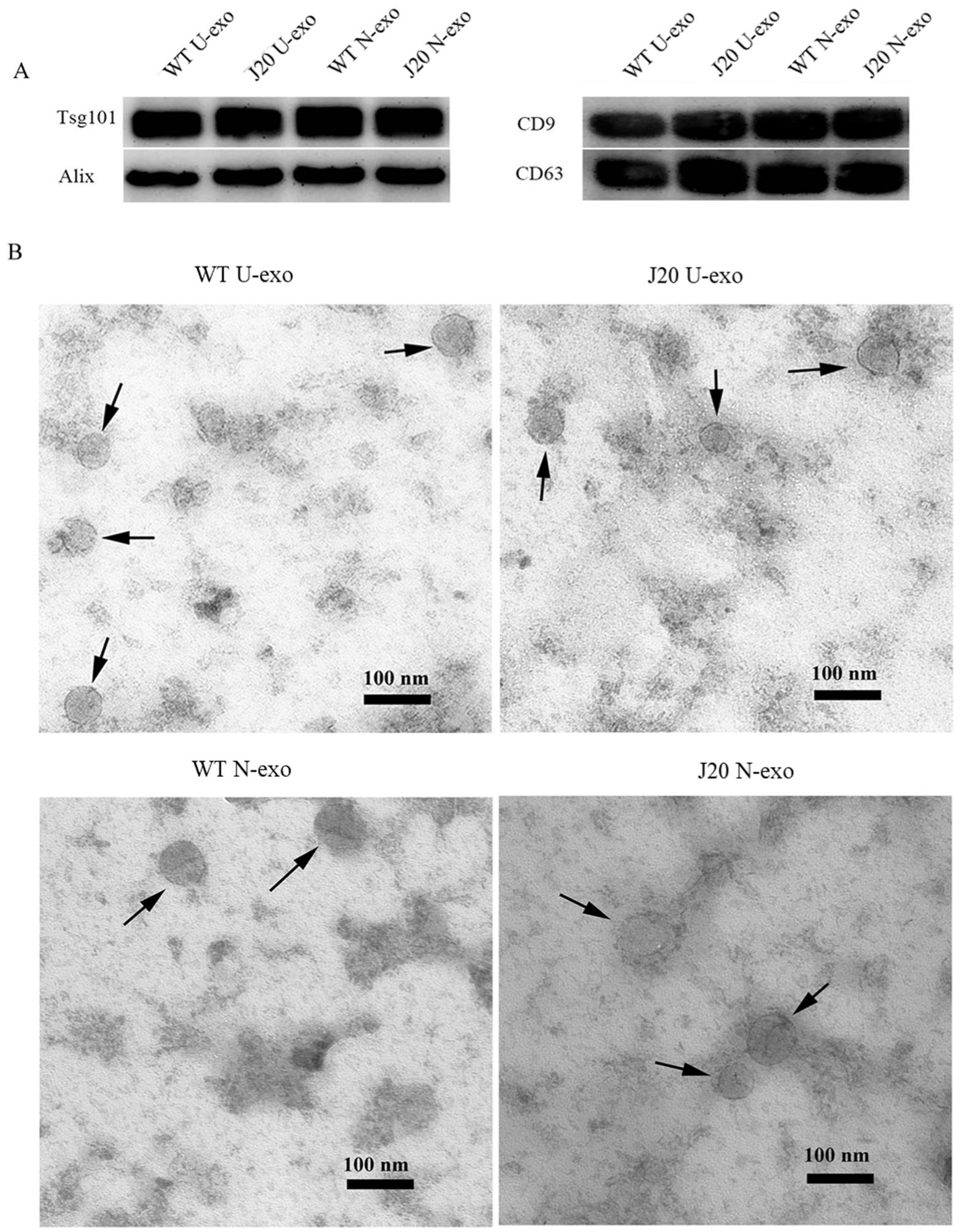
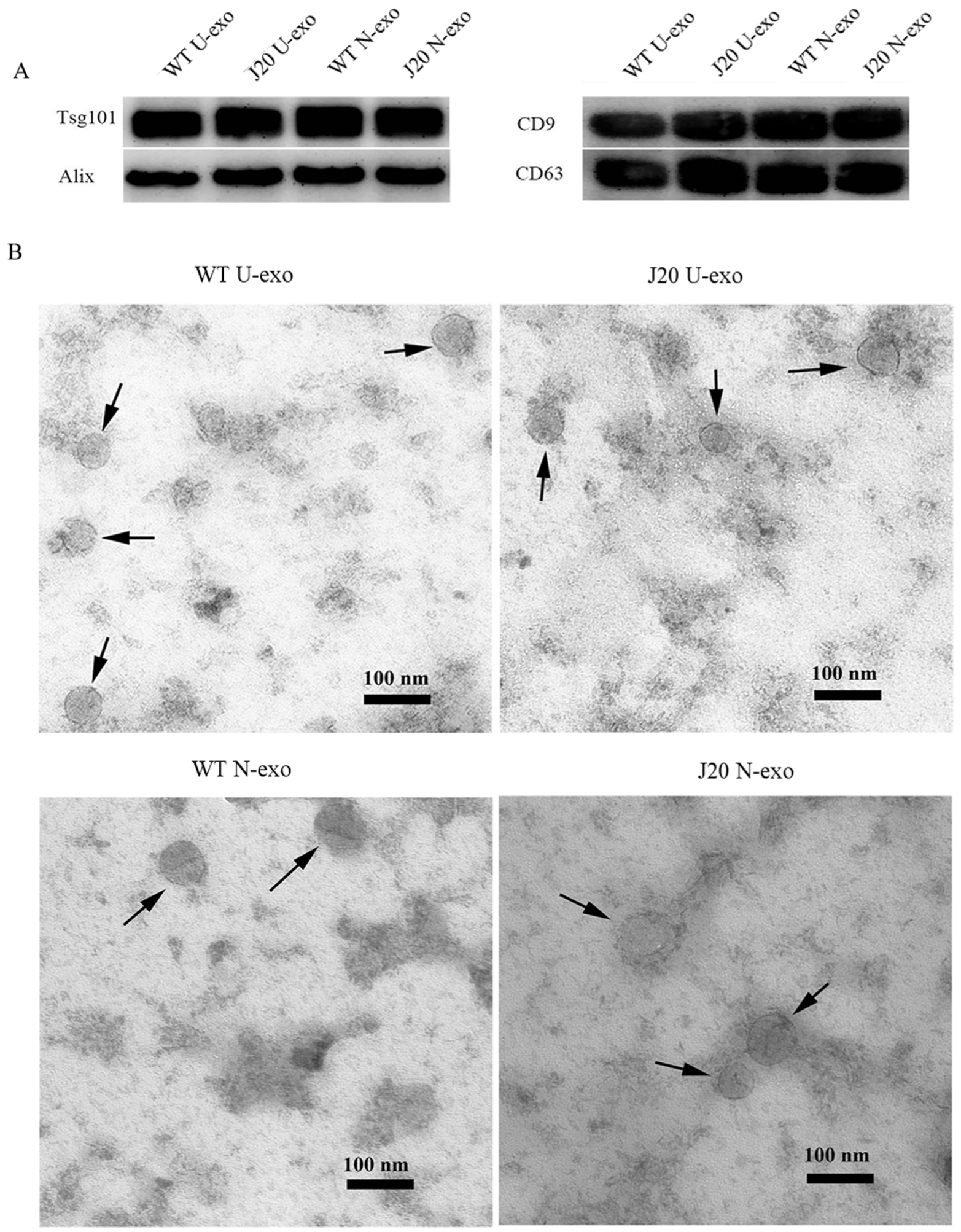
2.1. Characterization of Exosomes Isolated from Urine
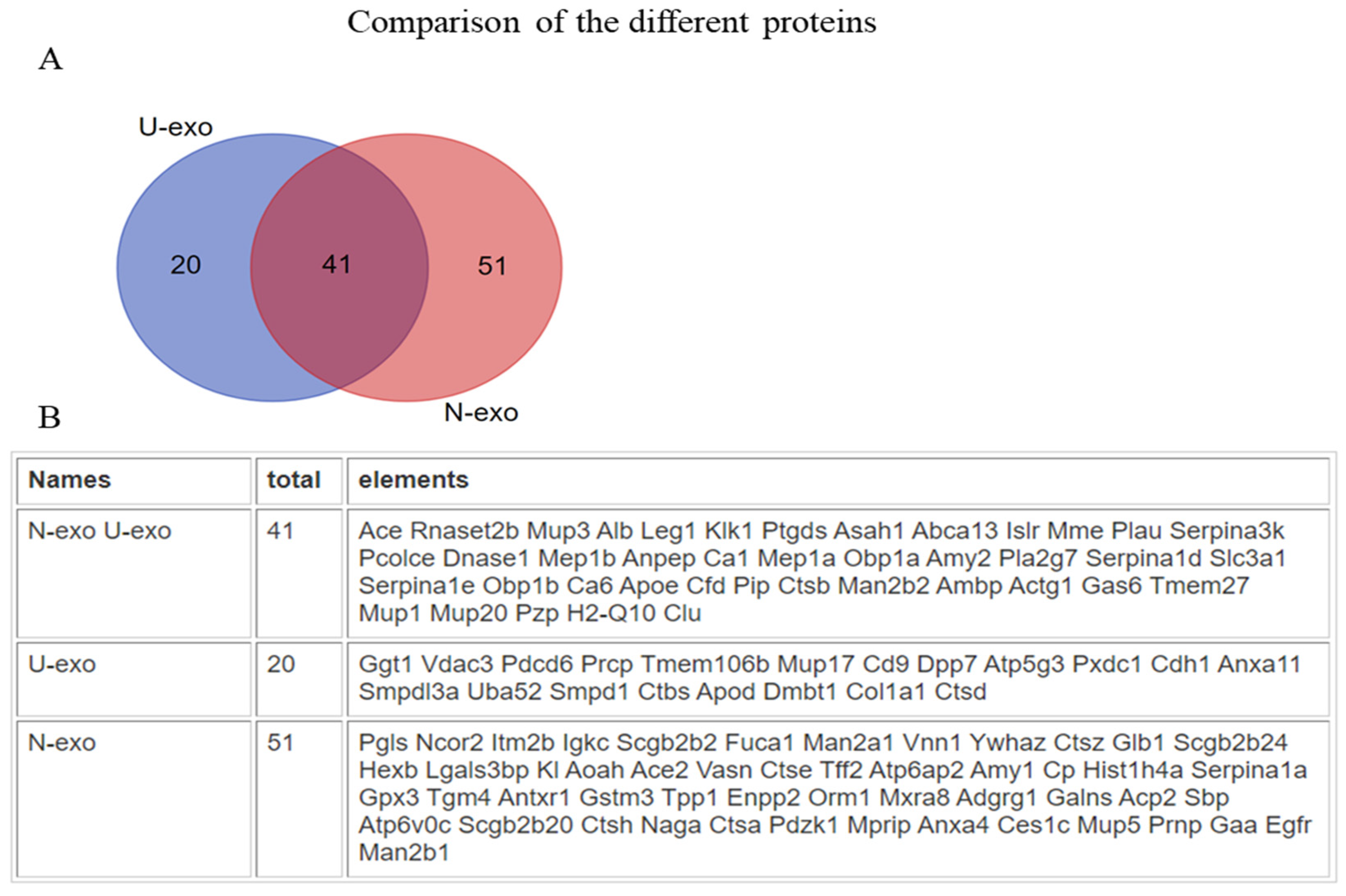
2.2. Analysis of the Exosome Proteins by Mass Spectrometry
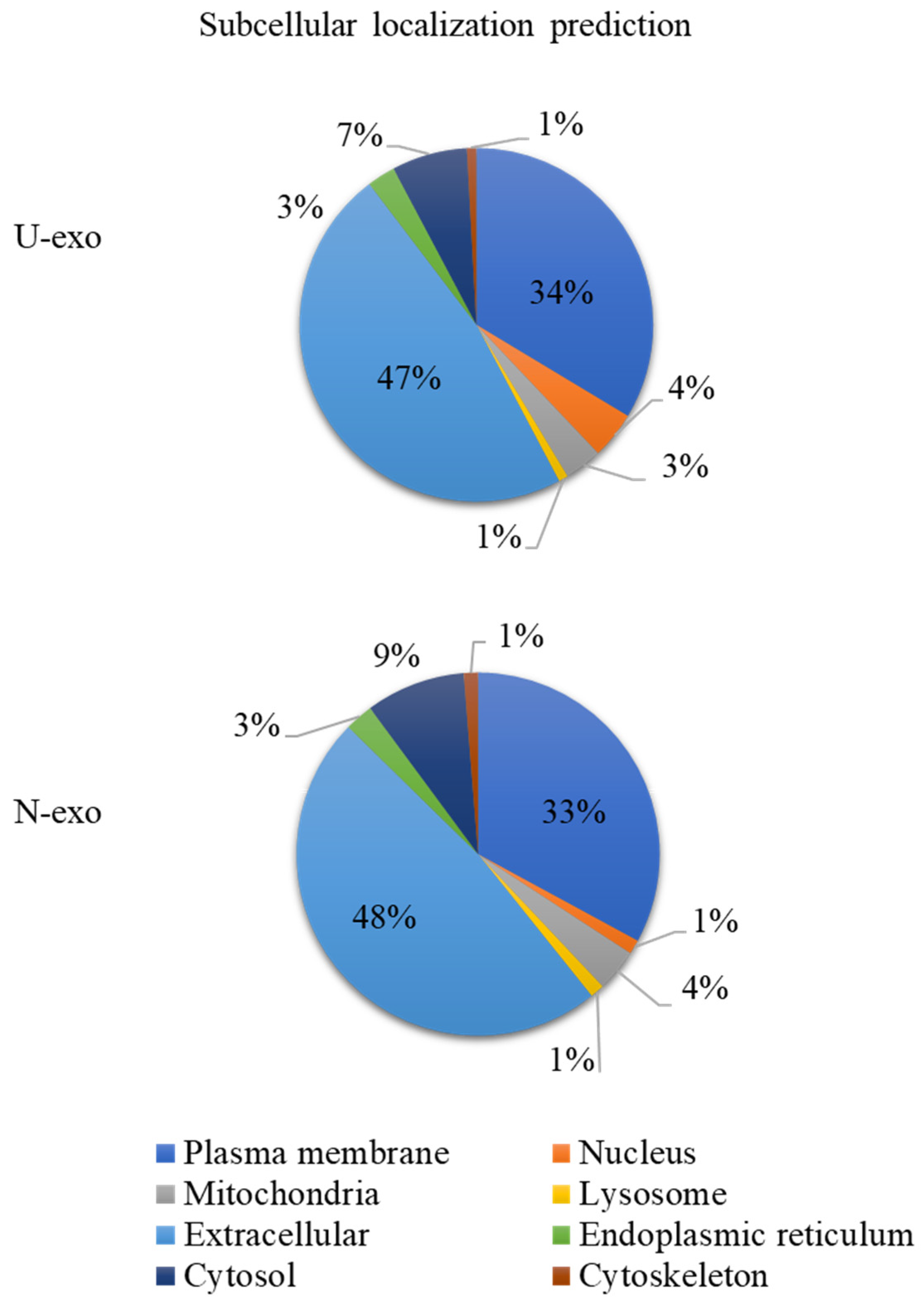
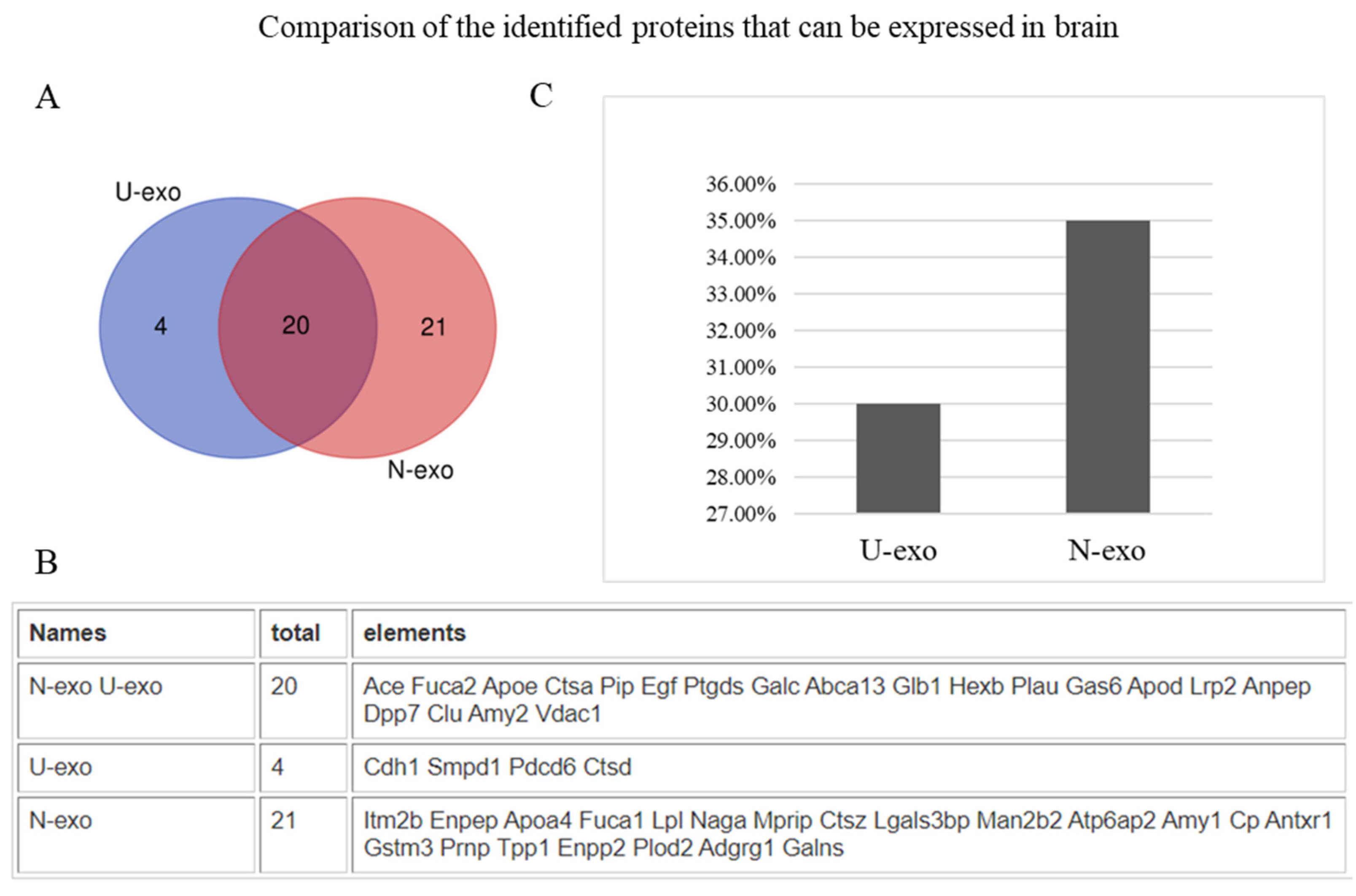
2.3. Analysis of Protein Subcellular Localization and Brain Expression
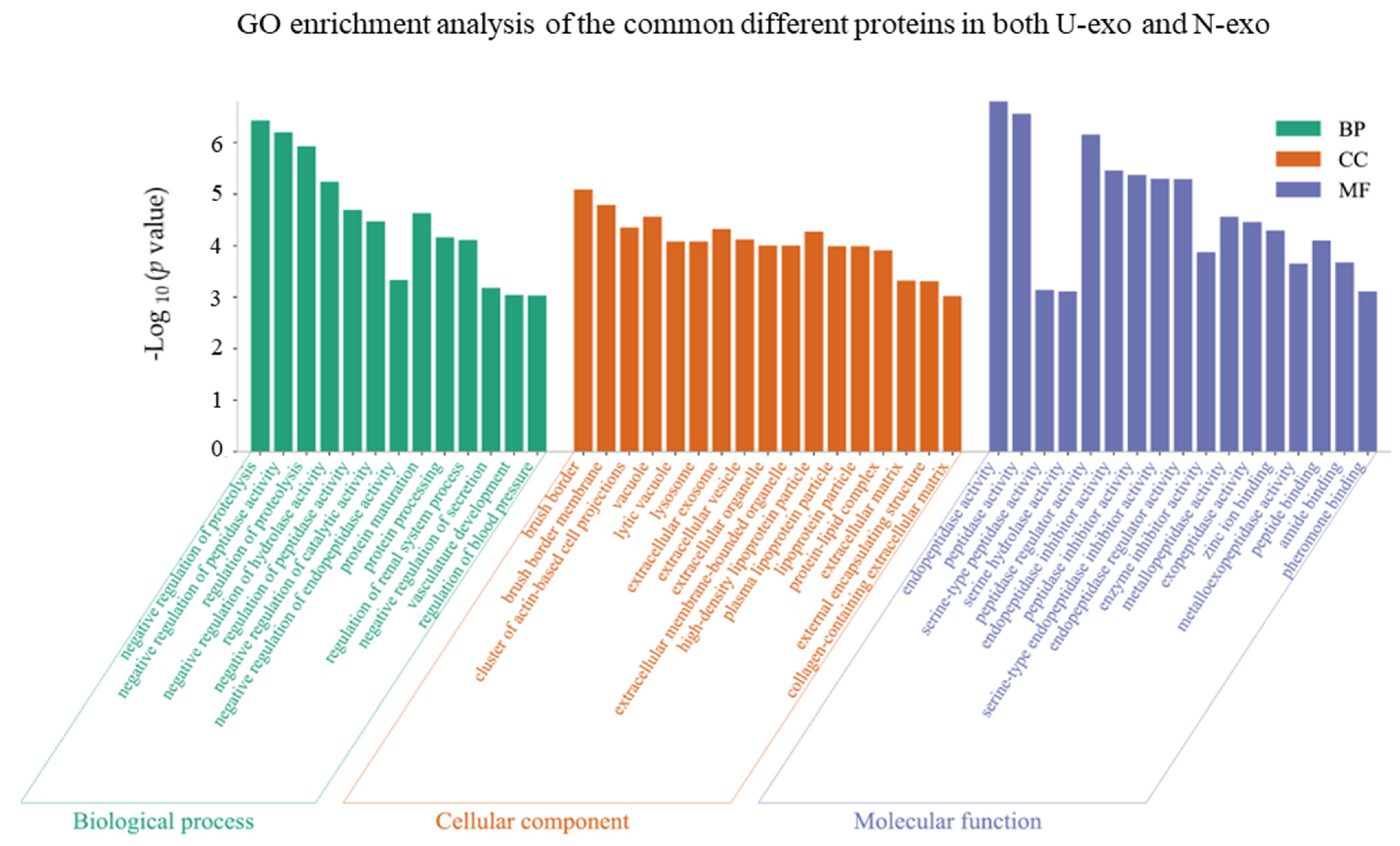
2.4. Gene Ontology (GO) Enrichment Analysis of the Common Different Exosome Proteins
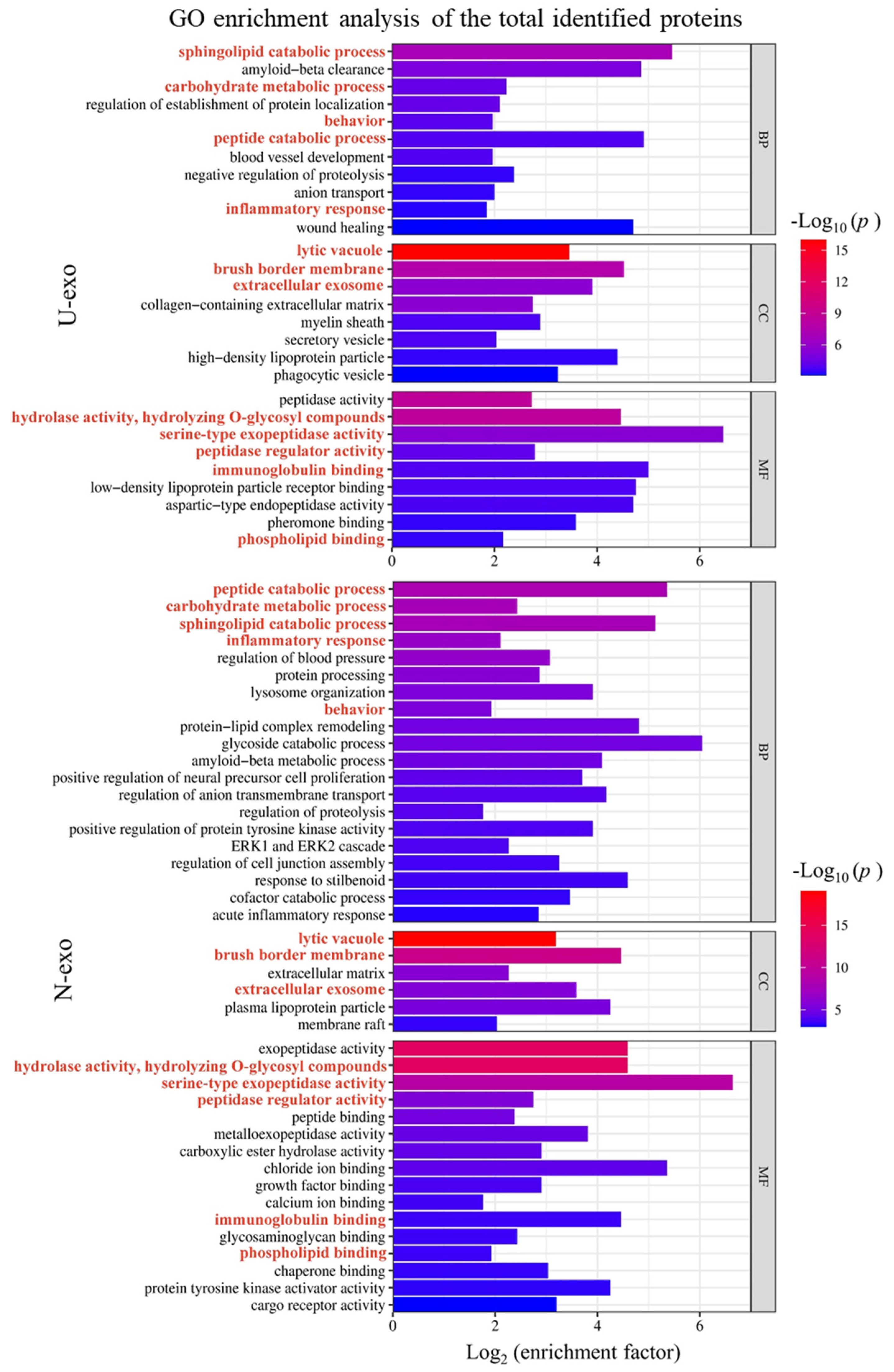
2.5. Gene Ontology (GO) Enrichment Analysis of Identified Exosome Proteins
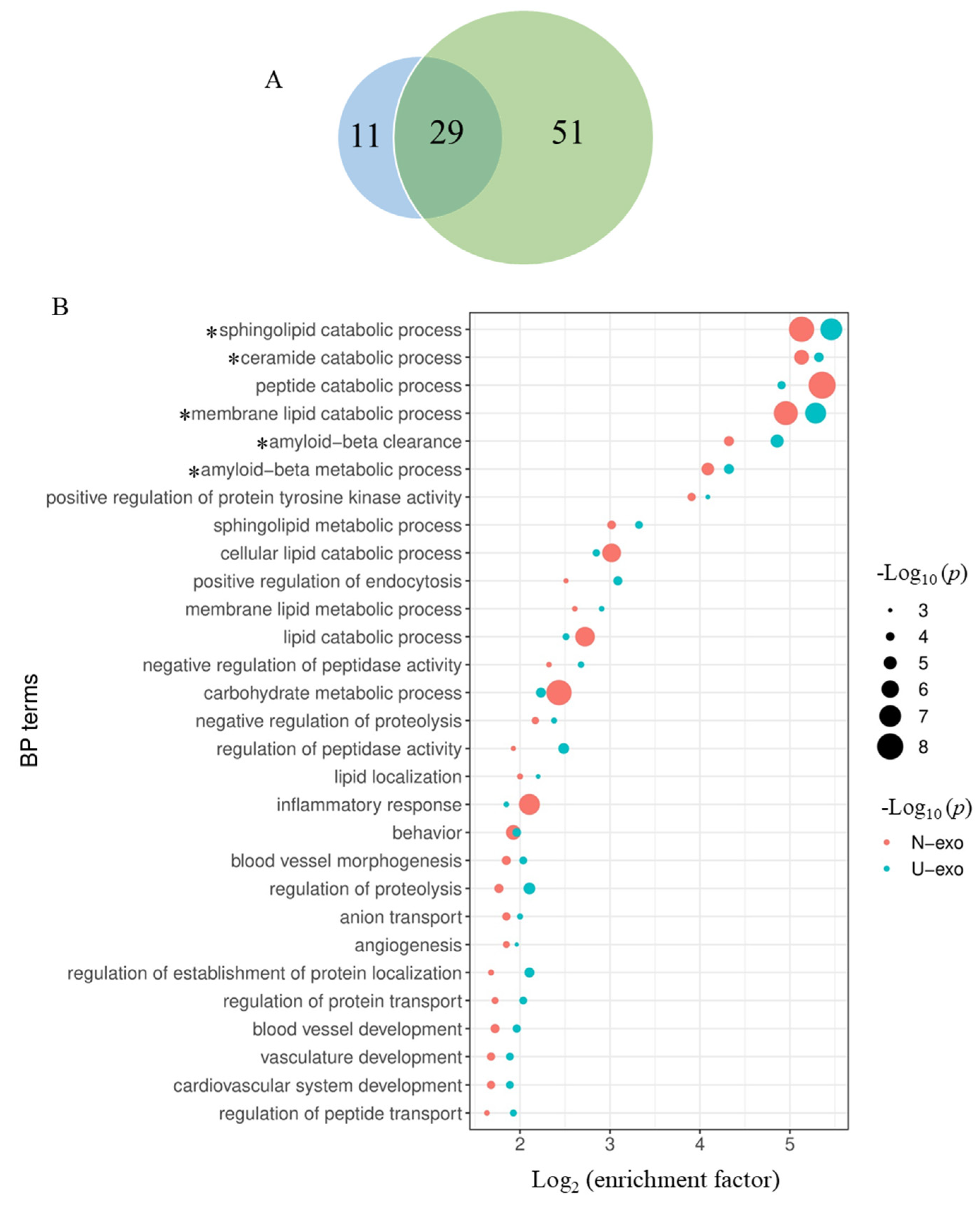
2.6. Comparison of the Enriched BP Terms by the Identified Proteins between U-exo and N-exo
2.7. Comparison of Exosome Proteins Participating in the Highly Enriched BP Terms in Both U-exo and N-exo
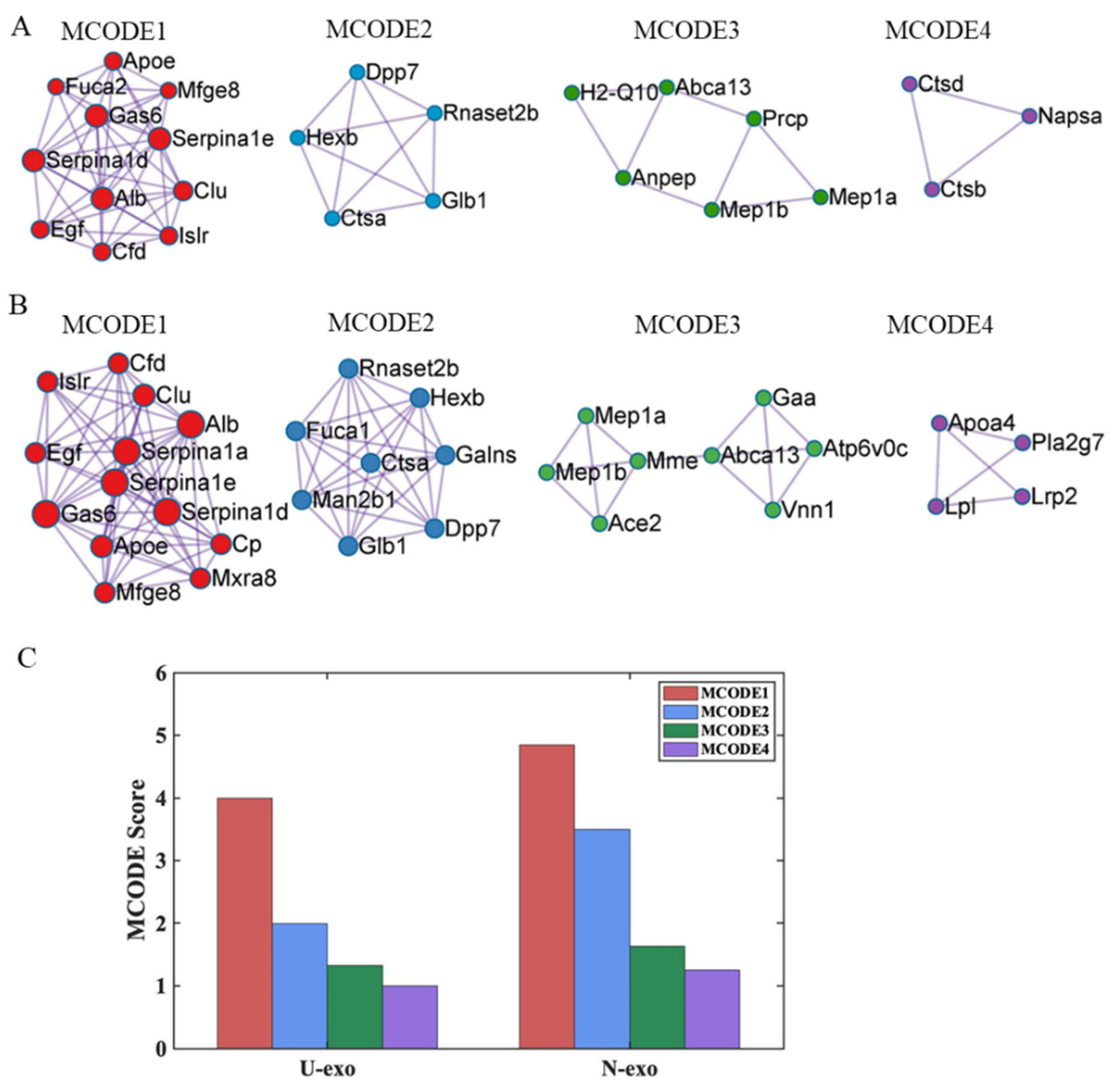
2.8. Protein–protein Interaction (PPI) Analysis
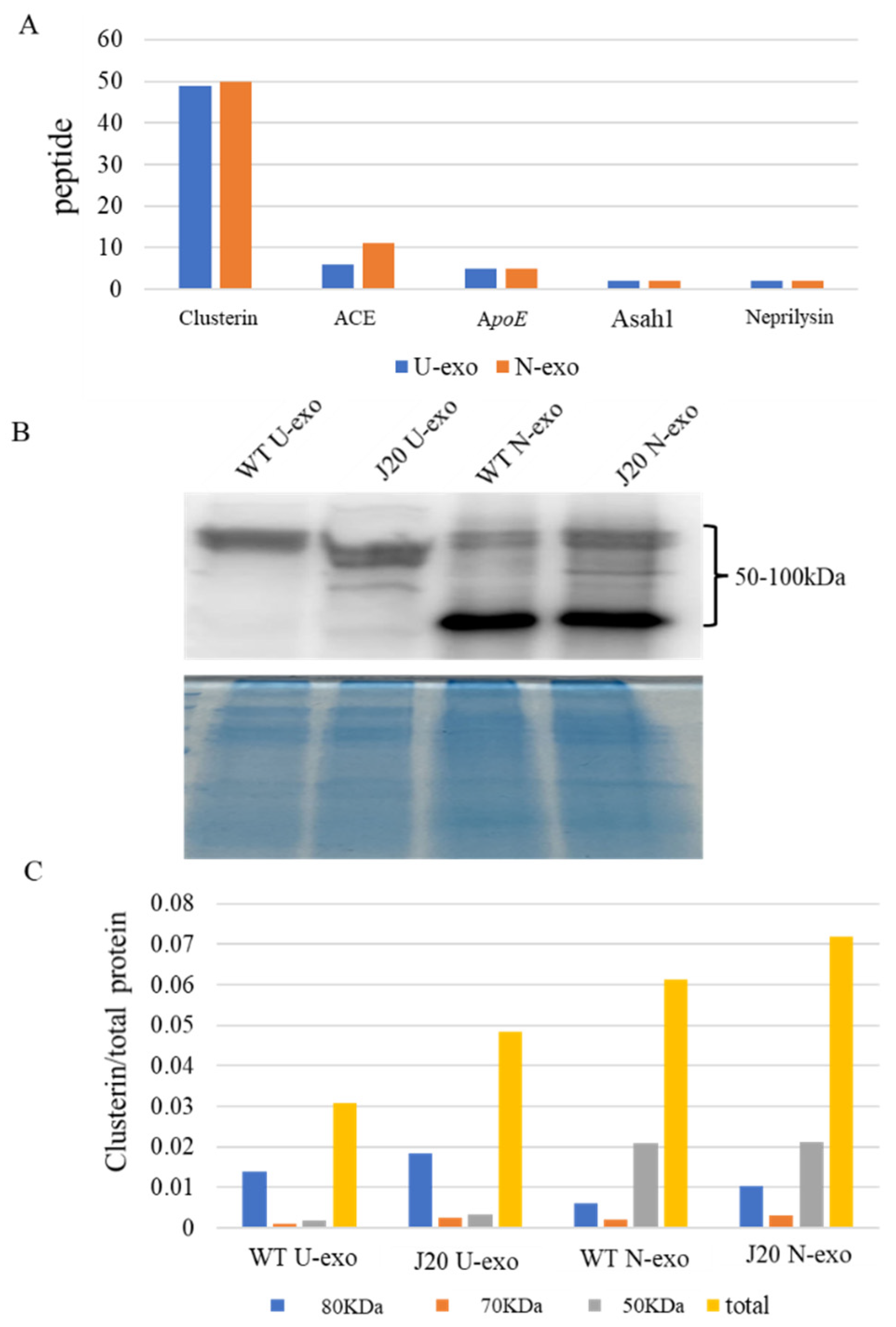
2.9. Quantification of Clusterin in U-exo and N-exo
3. Discussion
4. Materials and Methods
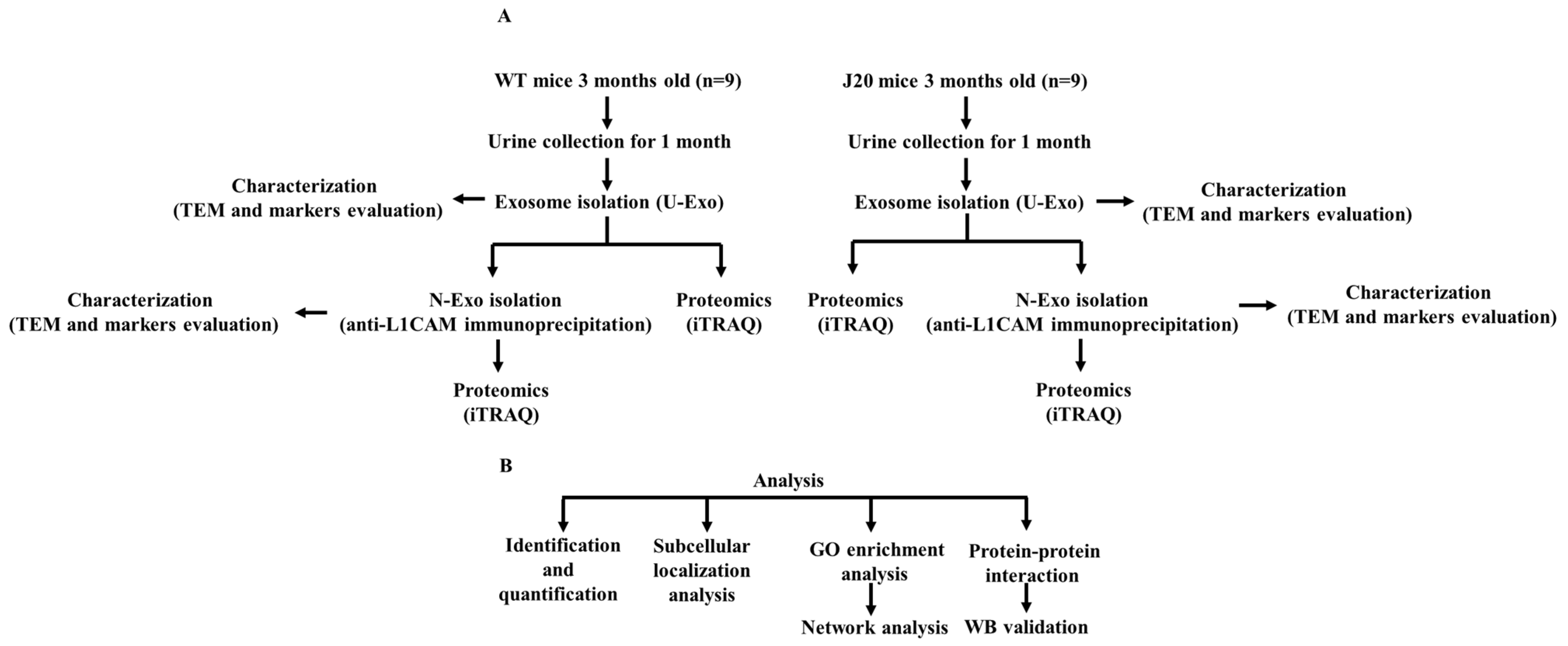
4.1. Animals and Samples
4.2. Exosome Isolation
4.3. Exosomal Protein Isolation
4.4. Western Blotting (WB)
4.5. Transmission Electron Microscopy (TEM)
4.6. Denaturation, Reduction, Cysteine Alkylation, Trypsin Digestion, iTRAQ Labeling, and Strong Cation Exchange Chromatography
4.7. Nano-LC Spotting
4.8. MALDI-TOF MS/MS Analysis
4.9. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent advancements in pathogenesis, diagnostics and treatment of alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of lipids in alzheimer’s disease pathology and potential therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Seo, Y.; Jo, Y.S.; Lee, S.; Cho, E.; Cazenave-Gassiot, A.; Shin, Y.-S.; Moon, M.H.; An, H.J.; Wenk, M.R.; et al. Brain lipidomics: From functional landscape to clinical significance. Sci. Adv. 2022, 8, eadc9317. [Google Scholar] [CrossRef]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar] [CrossRef]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408–413. [Google Scholar] [CrossRef]
- Natesan, V.; Kim, S.-J. Lipid Metabolism, Disorders and Therapeutic Drugs-Review. Biomol. Ther. 2021, 29, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.A.; Olzmann, J.A. Protein quality control and lipid droplet metabolism. Annu. Rev. Cell Dev. Biol. 2020, 36, 115–139. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Dysfunctional γ-Secretase in Familial Alzheimer’s Disease. Neurochem. Res. 2019, 44, 5–11. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-S.; Jo, S.A. Mechanisms of Amyloid-β Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. (Seoul) 2012, 20, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Armstrong, R.A. Classic beta-amyloid deposits cluster around large diameter blood vessels rather than capillaries in sporadic Alzheimer’s disease. Curr. Neurovasc. Res. 2006, 3, 289–294. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.; Ikezu, T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiol. Dis. 2019, 130, 104512. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, A.M.; Ikezu, T. Extracellular vesicle biology in alzheimer’s disease and related tauopathy. J. Neuroimmune Pharmacol. 2018, 13, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Schiapparelli, L.; Cline, H.T. Exosomes function in cell-cell communication during brain circuit development. Curr. Opin. Neurobiol. 2013, 23, 997–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Luarte, A.; Cisternas, P.; Caviedes, A.; Batiz, L.F.; Lafourcade, C.; Wyneken, U.; Henzi, R. Astrocytes at the Hub of the Stress Response: Potential Modulation of Neurogenesis by miRNAs in Astrocyte-Derived Exosomes. Stem Cells Int. 2017, 2017, 1719050. [Google Scholar] [CrossRef] [Green Version]
- Dalvi, P.; Sun, B.; Tang, N.; Pulliam, L. Immune activated monocyte exosomes alter microRNAs in brain endothelial cells and initiate an inflammatory response through the TLR4/MyD88 pathway. Sci. Rep. 2017, 7, 9954. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Chen, Z.; Zhang, M. Role of exosomes in the pathogenesis, diagnosis, and treatment of central nervous system diseases. J. Transl. Med. 2022, 20, 291. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Heidarzadeh, M.; Gürsoy-Özdemir, Y.; Kaya, M.; Eslami Abriz, A.; Zarebkohan, A.; Rahbarghazi, R.; Sokullu, E. Exosomal delivery of therapeutic modulators through the blood-brain barrier; promise and pitfalls. Cell Biosci. 2021, 11, 142. [Google Scholar] [CrossRef]
- Saeedi, S.; Israel, S.; Nagy, C.; Turecki, G. The emerging role of exosomes in mental disorders. Transl. Psychiatry 2019, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Bălașa, A.F.; Chircov, C.; Grumezescu, A.M. Body Fluid Biomarkers for Alzheimer’s Disease-An Up-To-Date Overview. Biomedicines 2020, 8, 421. [Google Scholar] [CrossRef] [PubMed]
- Rabbito, A.; Dulewicz, M.; Kulczyńska-Przybik, A.; Mroczko, B. Biochemical markers in alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011, 29, 26–32. [Google Scholar] [CrossRef] [Green Version]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Erbayraktar, Z.; Turan Evlice, A.; Yilmaz, G.; Yazici, C.; Yener, G.; Ulusu, N.N. Evaluation of Renal Function in Alzheimer’s Disease and Geriatric Patients: Results from a Turkish Two-center Study. J. Med. Biochem. 2017, 36, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Liu, Z.; Shen, Y.; Zhu, H. A novel perspective linkage between kidney function and alzheimer’s disease. Front. Cell. Neurosci. 2018, 12, 384. [Google Scholar] [CrossRef]
- Wu, J.-J.; Weng, S.-C.; Liang, C.-K.; Lin, C.-S.; Lan, T.-H.; Lin, S.-Y.; Lin, Y.-T. Effects of kidney function, serum albumin and hemoglobin on dementia severity in the oldest old people with newly diagnosed Alzheimer’s disease in a residential aged care facility: A cross-sectional study. BMC Geriatr. 2020, 20, 391. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.-C.; Russell, C.; Mitra, V.; Chung, R.; Hye, A.; Bazenet, C.; Lovestone, S.; Pike, I.; Ward, M. Glycosylation of human plasma clusterin yields a novel candidate biomarker of alzheimer’s disease. J. Proteome Res. 2015, 14, 5063–5076. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell. Mol. Life Sci. 2019, 76, 1833–1863. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.S.; Graham, L.; Uyar, A.; Preuss, C.; Howell, G.R.; Carter, G.W. Genetic perturbations of disease risk genes in mice capture transcriptomic signatures of late-onset Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 50. [Google Scholar] [CrossRef] [Green Version]
- Bali, J.; Gheinani, A.H.; Zurbriggen, S.; Rajendran, L. Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides. Proc. Natl. Acad. Sci. USA 2012, 109, 15307–15311. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic risk factors of sporadic alzheimer’s disease: Implications in the pathology, pathogenesis and treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [Green Version]
- Saganich, M.J.; Schroeder, B.E.; Galvan, V.; Bredesen, D.E.; Koo, E.H.; Heinemann, S.F. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J. Neurosci. 2006, 26, 13428–13436. [Google Scholar] [CrossRef] [Green Version]
- Cheng, I.H.; Scearce-Levie, K.; Legleiter, J.; Palop, J.J.; Gerstein, H.; Bien-Ly, N.; Puoliväli, J.; Lesné, S.; Ashe, K.H.; Muchowski, P.J.; et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007, 282, 23818–23828. [Google Scholar] [CrossRef] [Green Version]
- Mustapic, M.; Eitan, E.; Werner, J.K.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Front. Neurosci. 2017, 11, 278. [Google Scholar] [CrossRef]
- Sun, B.; Dalvi, P.; Abadjian, L.; Tang, N.; Pulliam, L. Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS 2017, 31, F9–F17. [Google Scholar] [CrossRef] [PubMed]
- Dagur, R.S.; Liao, K.; Sil, S.; Niu, F.; Sun, Z.; Lyubchenko, Y.L.; Peeples, E.S.; Hu, G.; Buch, S. Neuronal-derived extracellular vesicles are enriched in the brain and serum of HIV-1 transgenic rats. J. Extracell. Vesicles 2020, 9, 1703249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatr. 2020, 91, 720–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Elahi, F.M.; Mustapic, M.; Kapogiannis, D.; Pryhoda, M.; Gilmore, A.; Gorgens, K.A.; Davidson, B.; Granholm, A.-C.; Ledreux, A. Altered levels of plasma neuron-derived exosomes and their cargo proteins characterize acute and chronic mild traumatic brain injury. FASEB J. 2019, 33, 5082–5088. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Leoutsakos, J.-M.; Troncoso, J.; Lyketsos, C.G.; Oh, E.S.; Kapogiannis, D. Neuronal-Derived EV Biomarkers Track Cognitive Decline in Alzheimer’s Disease. Cells 2022, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kim, S.; Su, Y.; Sharma, M.; Kumar, P.; Singh, S.; Lee, J.; Furdui, C.M.; Singh, R.; Hsu, F.-C.; et al. Brain cell-derived exosomes in plasma serve as neurodegeneration biomarkers in male cynomolgus monkeys self-administrating oxycodone. EBioMedicine 2021, 63, 103192. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Pei, F.; Zeng, C.; Yao, Y.; Liao, W.; Zhao, Z. Extracellular vesicles in liquid biopsies: Potential for disease diagnosis. Biomed. Res. Int. 2021, 2021, 6611244. [Google Scholar] [CrossRef]
- Jan, A.T.; Rahman, S.; Khan, S.; Tasduq, S.A.; Choi, I. Biology, pathophysiological role, and clinical implications of exosomes: A critical appraisal. Cells 2019, 8, 99. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Regan, B.; Owen, K.; Collins, D.; McEneaney, D.; Megson, I.L.; Redmond, E.M.; Cahill, P.A. Exosomal Composition, Biogenesis and Profiling Using Point-of-Care Diagnostics-Implications for Cardiovascular Disease. Front. Cell Dev. Biol. 2022, 10, 853451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Chen, Q.; Lin, L.; Sha, C.; Li, T.; Liu, Y.; Yin, X.; Xu, Y.; Chen, L.; Gao, W.; et al. Regulation of exosome production and cargo sorting. Int. J. Biol. Sci. 2021, 17, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Boriachek, K.; Islam, M.N.; Möller, A.; Salomon, C.; Nguyen, N.-T.; Hossain, M.S.A.; Yamauchi, Y.; Shiddiky, M.J.A. Biological functions and current advances in isolation and detection strategies for exosome nanovesicles. Small 2018, 14, 1702153. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, S.; Öner, P. Could low α-N-acetylgalactosaminidase plasma concentration cause schizophrenia? World J. Biol. Psychiatry 2022, 1–8. [Google Scholar] [CrossRef]
- Korlimarla, A.; Lim, J.-A.; Kishnani, P.S.; Sun, B. An emerging phenotype of central nervous system involvement in Pompe disease: From bench to bedside and beyond. Ann. Transl. Med. 2019, 7, 289. [Google Scholar] [CrossRef]
- Cui, Y.; Huang, M.; He, Y.; Zhang, S.; Luo, Y. Genetic ablation of apolipoprotein A-IV accelerates Alzheimer’s disease pathogenesis in a mouse model. Am. J. Pathol. 2011, 178, 1298–1308. [Google Scholar] [CrossRef]
- Zhu, C.; Aguzzi, A. Prion protein and prion disease at a glance. J. Cell Sci. 2021, 134, jcs245605. [Google Scholar] [CrossRef]
- Doody, R.S.; Demirovic, J.; Ballantyne, C.M.; Chan, W.; Barber, R.; Powell, S.; Pavlik, V. Texas Alzheimer’s Disease Research and Care Consortium Lipoprotein-associated phospholipase A2, homocysteine, and Alzheimer’s disease. Alzheimers Dement. (Amst.) 2015, 1, 464–471. [Google Scholar] [CrossRef]
- Lemon, N.; Canepa, E.; Ilies, M.A.; Fossati, S. Carbonic anhydrases as potential targets against neurovascular unit dysfunction in alzheimer’s disease and stroke. Front. Aging Neurosci. 2021, 13, 772278. [Google Scholar] [CrossRef] [PubMed]
- Owlett, L.D.; Karaahmet, B.; Le, L.; Belcher, E.K.; Dionisio-Santos, D.; Olschowka, J.A.; Elliott, M.R.; O’Banion, M.K. Gas6 induces inflammation and reduces plaque burden but worsens behavior in a sex-dependent manner in the APP/PS1 model of Alzheimer’s disease. J. Neuroinflammation 2022, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Marengo, L.; Armbrust, F.; Schoenherr, C.; Storck, S.E.; Schmitt, U.; Zampar, S.; Wirths, O.; Altmeppen, H.; Glatzel, M.; Kaether, C.; et al. Meprin β knockout reduces brain Aβ levels and rescues learning and memory impairments in the APP/lon mouse model for Alzheimer’s disease. Cell. Mol. Life Sci. 2022, 79, 168. [Google Scholar] [CrossRef] [PubMed]
- El-Amouri, S.S.; Zhu, H.; Yu, J.; Marr, R.; Verma, I.M.; Kindy, M.S. Neprilysin: An enzyme candidate to slow the progression of Alzheimer’s disease. Am. J. Pathol. 2008, 172, 1342–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hook, V.; Yoon, M.; Mosier, C.; Ito, G.; Podvin, S.; Head, B.P.; Rissman, R.; O’Donoghue, A.J.; Hook, G. Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140428. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Nixon, R.A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl.Acad. Sci. USA 1990, 87, 3861–3865. [Google Scholar] [CrossRef] [Green Version]
- Oberstein, T.J.; Utz, J.; Spitzer, P.; Klafki, H.W.; Wiltfang, J.; Lewczuk, P.; Kornhuber, J.; Maler, J.M. The role of cathepsin B in the degradation of aβ and in the production of aβ peptides starting with ala2 in cultured astrocytes. Front. Mol. Neurosci. 2020, 13, 615740. [Google Scholar] [CrossRef]
- Hook, V.; Funkelstein, L.; Wegrzyn, J.; Bark, S.; Kindy, M.; Hook, G. Cysteine Cathepsins in the secretory vesicle produce active peptides: Cathepsin L generates peptide neurotransmitters and cathepsin B produces beta-amyloid of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1824, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.-P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and alzheimer’s disease: From pathogenesis to therapeutic implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.; DiGiacomo, P.; Born, D.E.; Georgiadis, M.; Zeineh, M. Iron and alzheimer’s disease: From pathology to imaging. Front. Hum. Neurosci. 2022, 16, 838692. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, J.; Shen, Y.; Li, H.; Rausch, W.-D.; Huang, X. Iron dyshomeostasis and ferroptosis: A new alzheimer’s disease hypothesis? Front. Aging Neurosci. 2022, 14, 830569. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-S.; Zhang, L.-H.; Yu, P.-P.; Gou, Y.-J.; Zhao, J.; You, L.-H.; Wang, Z.-Y.; Zheng, X.; Yan, L.-J.; Yu, P.; et al. Ceruloplasmin, a potential therapeutic agent for alzheimer’s disease. Antioxid. Redox Signal. 2018, 28, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Okino, N.; He, X.; Gatt, S.; Sandhoff, K.; Ito, M.; Schuchman, E.H. The reverse activity of human acid ceramidase. J. Biol. Chem. 2003, 278, 29948–29953. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Okino, N.; Dhami, R.; Dagan, A.; Gatt, S.; Schulze, H.; Sandhoff, K.; Schuchman, E.H. Purification and characterization of recombinant, human acid ceramidase. Catalytic reactions and interactions with acid sphingomyelinase. J. Biol. Chem. 2003, 278, 32978–32986. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huang, H.; Ding, S.-F. Sphingosine-1-phosphate promotes the proliferation and attenuates apoptosis of Endothelial progenitor cells via S1PR1/S1PR3/PI3K/Akt pathway. Cell Biol. Int. 2018, 42, 1492–1502. [Google Scholar] [CrossRef]
- Bonnaud, S.; Niaudet, C.; Legoux, F.; Corre, I.; Delpon, G.; Saulquin, X.; Fuks, Z.; Gaugler, M.-H.; Kolesnick, R.; Paris, F. Sphingosine-1-phosphate activates the AKT pathway to protect small intestines from radiation-induced endothelial apoptosis. Cancer Res. 2010, 70, 9905–9915. [Google Scholar] [CrossRef]
- Long, H.-Z.; Cheng, Y.; Zhou, Z.-W.; Luo, H.-Y.; Wen, D.-D.; Gao, L.-C. PI3K/AKT signal pathway: A target of natural products in the prevention and treatment of alzheimer’s disease and parkinson’s disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef] [PubMed]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Zang, G.; Fang, L.; Chen, L.; Wang, C. Ameliorative effect of nicergoline on cognitive function through the PI3K/AKT signaling pathway in mouse models of Alzheimer’s disease. Mol. Med. Report. 2018, 17, 7293–7300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, S.; Govindarajulu, M.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Autotaxin−lysophosphatidic acid signaling in alzheimer’s disease. Int. J. Mol. Sci. 2018, 19, 1827. [Google Scholar] [CrossRef] [Green Version]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Role of clusterin in the brain vascular clearance of amyloid-β. Proc. Natl. Acad. Sci. USA 2017, 114, 8681–8682. [Google Scholar] [CrossRef] [Green Version]
- Hemming, M.L.; Selkoe, D.J. Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J. Biol. Chem. 2005, 280, 37644–37650. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N.S. Protein-protein interaction detection: Methods and analysis. Int. J. Proteomics 2014, 2014, 147648. [Google Scholar] [CrossRef]
- Papathanasiou, I.; Balis, C.; Trachana, V.; Mourmoura, E.; Tsezou, A. The synergistic function of miR-140-5p and miR-146a on TLR4-mediated cytokine secretion in osteoarthritic chondrocytes. Biochem. Biophys. Res. Commun. 2020, 522, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Meng, L.-B.; Hao, M.-L.; Yang, J.-F.; Zou, T. Identification of Co-expressed Genes between Atrial Fibrillation and Stroke. Front. Neurol. 2020, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, R.; Okuno, A.; Yoshimi, T.; Watanabe, A.; Maruyama, M.; Takikawa, O. Partial Identification of Amyloid-β Degrading Activity in Human Serum. Nagoya J. Med. Sci. 2019, 81, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, S.Y.; Lim, S.; Choi, H.R.; Choi, Y.; Kim, M.; Kim, S.; Lee, Y.; Han, K.H.; Chung, W.-S.; et al. Anti-inflammatory clearance of amyloid-β by a chimeric Gas6 fusion protein. Nat. Med. 2022, 28, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Guo, C. Albumin Alters the Conformational Ensemble of Amyloid-β by Promiscuous Interactions: Implications for Amyloid Inhibition. Front. Mol. Biosci. 2020, 7, 629520. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, X.; Song, L.; Yuan, F.; Yan, Y. Matrix Remodeling-Associated Protein 8 as a Novel Indicator Contributing to Glioma Immune Response by Regulating Ferroptosis. Front. Immunol. 2022, 13, 834595. [Google Scholar] [CrossRef]
- van Duijn, S.; Bulk, M.; van Duinen, S.G.; Nabuurs, R.J.A.; van Buchem, M.A.; van der Weerd, L.; Natté, R. Cortical Iron Reflects Severity of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.W.; Wong, C.H.Y. Aging- and vascular-related pathologies. Microcirculation 2019, 26, e12463. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Alzheimer’s Disease Neuroimaging Initiative Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.-I.; Maniwa, T.; Tanaka, T.; Satoh, K.; Okunishi, H.; Oda, T. Proteomic analysis of calcified abdominal and thoracic aortic aneurysms. Int. J. Mol. Med. 2012, 30, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Satoh, K.; Maniwa, T.; Oda, T.; Matsumoto, K.-I. Proteomic profiling for the identification of serum diagnostic biomarkers for abdominal and thoracic aortic aneurysms. Proteome Sci. 2013, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Satoh, K.; Maniwa, T.; Matsumoto, K.-I. Proteomic analysis for the identification of serum diagnostic markers for joint hypermobility syndrome. Int. J. Mol. Med. 2016, 37, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.-I. Phosphorylation of extracellular matrix tenascin-X detected by differential mass tagging followed by nanoLC-MALDI-TOF/TOF-MS/MS using ProteinPilot software. Connect. Tissue Res. 2012, 53, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C. Modelling patterns of agreement for nominal scales. Stat. Med. 2008, 27, 810–830. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unused ProtScore a | % Coverage b | Peptides (95%) c | UniProt Accession | Gene Symbol | Name | Mean Ratio d |
|---|---|---|---|---|---|---|
| 3.15 | 9.5 | 1 | Q80 × 71 | Tmem106b | Transmembrane protein 106B | 2.6911 |
| 14 | 47.2 | 8 | Q9D3H2 | Obp1a | Odorant-binding protein 1a | 2.5735 |
| 6.38 | 17.4 | 3 | P18761 | Ca6 | Carbonic anhydrase 6 | 2.4163 |
| 2.35 | 15.6 | 1 | Q8JZU6 | Pxdc1 | PX domain-containing protein 1 | 2.4042 |
| 8 | 17.2 | 4 | P13634 | Ca1 | Carbonic anhydrase 1 | 2.3034 |
| 6.54 | 32.2 | 4 | A2AEP0 | Obp1b | Odorant-binding protein 1b | 2.2020 |
| 4.26 | 12.3 | 2 | P01898 | H2-Q10 | H-2 class I histocompatibility antigen, Q10 alpha chain | 1.9817 |
| 89.56 | 79.1 | 75 | P00688 | Amy2 | Pancreatic alpha-amylase | 1.9728 |
| 44.52 | 48.7 | 23 | P07724 | Alb | Serum albumin | 1.9404 |
| 5.49 | 13.4 | 2 | Q60963 | Pla2g7 | Platelet-activating factor acetylhydrolase | 1.8847 |
| 19.33 | 39 | 10 | Q07456 | Ambp | Protein AMBP | 1.8396 |
| 14.02 | 11.4 | 8 | Q60997 | Dmbt1 | Deleted in malignant brain tumors 1 protein | 1.8363 |
| 10.55 | 54.5 | 24 | Q00898 | Serpina1e | Alpha-1-antitrypsin 1-5 | 1.8309 |
| 15.5 | 36.5 | 11 | Q8C6C9 | Leg1 | Protein LEG1 homolog | 1.8007 |
| 3.52 | 87.2 | 371 | P11588 | Mup1 | Major urinary protein 1 | 1.7617 |
| 43.75 | 63 | 32 | Q00897 | Serpina1d | Alpha-1-antitrypsin 1-4 | 1.7579 |
| 14.5 | 34.5 | 10 | P07759 | Serpina3k | Serine protease inhibitor A3K | 1.7509 |
| 13.25 | 89.5 | 116 | Q5FW60 | Mup20 | Major urinary protein 20 | 1.6810 |
| 4 | 6.8 | 2 | Q61391 | Mme | Neprilysin | 1.6804 |
| 10.8 | 52.7 | 5 | P02816 | Pip | Prolactin-inducible protein homolog | 1.6792 |
| 4.7 | 22.4 | 2 | P10605 | Ctsb | Cathepsin B | 1.6491 |
| 2.18 | 6.8 | 1 | Q6GU68 | Islr | Immunoglobulin superfamily containing leucine-rich repeat protein | 1.6337 |
| 5.4 | 30.2 | 4 | P51910 | Apod | Apolipoprotein D | 1.5999 |
| 14.96 | 17.2 | 6 | P09470 | Ace | Angiotensin-converting enzyme | 1.5564 |
| 9.02 | 9.2 | 4 | P56384 | Atp5g3 | ATP synthase F(0) complex subunit C3 | 1.5092 |
| 9.02 | 9.2 | 4 | Q5SSE9 | Abca13 | ATP-binding cassette sub-family A member 13 | 1.5018 |
| 21.68 | 40.6 | 21 | P15947 | Klk1 | Kallikrein-1 | 1.4949 |
| 4.38 | 11.2 | 2 | C0HKG6 | Rnaset2b | Ribonuclease T2-B | 1.4947 |
| 2 | 10.2 | 1 | P06869 | Plau | Urokinase-type plasminogen activator | 1.4821 |
| 4 | 27.9 | 3 | Q60931 | Vdac3 | Voltage-dependent anion-selective channel protein 3 | 1.4567 |
| 4 | 4.4 | 2 | O54782 | Man2b2 | Epididymis-specific alpha-mannosidase | 1.4522 |
| 6.22 | 14.9 | 3 | P97384 | Anxa11 | Annexin A11 | 1.4433 |
| 10.11 | 19.3 | 5 | P08226 | Apoe | Apolipoprotein E | 1.4288 |
| 12.42 | 9.8 | 5 | Q61838 | Pzp | Pregnancy zone protein | 1.4280 |
| 18.4 | 46.1 | 12 | P49183 | Dnase1 | Deoxyribonuclease-1 | 1.4277 |
| 3.53 | 12.4 | 3 | P18242 | Ctsd | Cathepsin D | 1.4262 |
| 12.14 | 32.8 | 7 | O09114| | Ptgds | Prostaglandin-H2 D-isomerase | 1.4217 |
| 4.26 | 7.1 | 2 | Q61592 | Gas6 | Growth arrest-specific protein 6 | 1.3947 |
| 4.07 | 9.6 | 2 | Q8R242 | Ctbs | Di-N-acetylchitobiase | 1.3938 |
| 9 | 12.4 | 4 | P70158 | Smpdl3a | Acid sphingomyelinase-like phosphodiesterase 3a | 1.3852 |
| 133.09 | 88 | 230 | P04939 | Mup3 | Major urinary protein 3 | 1.3838 |
| 12.55 | 19.2 | 8 | Q61847 | Mep1b | Meprin A subunit beta | 1.3830 |
| 3.71 | 15.5 | 2 | Q9WV54 | Asah1 | Acid ceramidase | 1.3824 |
| 0.8 | 5.3 | 0 | P40240 | Cd9 | CD9 antigen | 1.3763 |
| 23 | 87.2 | 367 | B5X0G2 | Mup17 | Major urinary protein 17 | 1.3637 |
| 8.11 | 10.2 | 4 | P09803 | Cdh1 | Cadherin-1 | 1.3598 |
| 24.08 | 47.1 | 17 | P03953 | Cfd | Complement factor D | 1.3478 |
| 4.04 | 12.6 | 2 | P12815 | Pdcd6 | Programmed cell death protein 6 | 1.3386 |
| 2.01 | 9.6 | 1 | Q04519 | Smpd1 | Sphingomyelin phosphodiesterase | 1.3335 |
| 13.79 | 27.5 | 7 | P63260 | Actg1 | Actin, cytoplasmic 2 | 1.3288 |
| 60.93 | 60.7 | 49 | Q06890 | Clu | Clusterin | 1.3055 |
| 2.2 | 11.7 | 1 | Q9ESG4 | Tmem27 | Collectrin | 1.3012 |
| 23.72 | 33 | 15 | Q9ET22 | Dpp7 | Dipeptidyl peptidase 2 | 1.2554 |
| 16.26 | 22.7 | 8 | Q60928 | Ggt1 | Gamma-glutamyltranspeptidase 1 | 1.2462 |
| 14.17 | 21.3 | 7 | Q91WV7 | Slc3a1 | Neutral and basic amino acid transport protein rBAT | 1.2417 |
| 12.29 | 13.9 | 7 | P97449 | Anpep | Aminopeptidase N | 1.2401 |
| 4 | 15.2 | 2 | Q61398 | Pcolce | Procollagen C-endopeptidase enhancer 1 | 1.2320 |
| 8 | 14.5 | 4 | Q7TMR0 | Prcp | Lysosomal Pro-X carboxypeptidase | 1.2307 |
| 148.27 | 56.1 | 175 | P28825 | Mep1a | Meprin A subunit alpha | 1.2239 |
| 10.59 | 20.4 | 5 | P11087 | Col1a1 | Collagen alpha-1(I) chain | 1.2221 |
| 6.01 | 44.5 | 3 | P62984 | Uba52 | Ubiquitin-60S ribosomal protein L40 | 1.2113 |
| Unused ProtScore a | % Coverage b | UniProt Accession | Gene Symbol | Name | Peptides (95%) c | 115:114 |
|---|---|---|---|---|---|---|
| 6.38 | 39.5 | Q6UGQ3 | Scgb2b2 | Secretoglobin family 2B member 2 | 3 | 3.6573 |
| 14.09 | 48.5 | Q9D3H2 | Obp1a | Odorant-binding protein 1a | 11 | 2.9136 |
| 7.74 | 8.7 | Q8BZH1 | Tgm4 | Protein-glutamine gamma-glutamyltransferase 4 | 4 | 2.7917 |
| 2.01 | 4.5 | Q9WU42 | Ncor2 | Nuclear receptor corepressor 2 | 1 | 2.7166 |
| 4 | 17 | Q7M747 | Scgb2b24 | Secretoglobin family 2B member 24 | 2 | 2.4941 |
| 4 | 15.1 | P18761 | Ca6 | Carbonic anhydrase 6 | 3 | 2.4084 |
| 4 | 24 | A2AEP0 | Obp1b | Odorant-binding protein 1b | 4 | 2.4014 |
| 5.42 | 66.1 | P00687 | Amy1 | Alpha-amylase 1 | 36 | 2.3926 |
| 4.28 | 10.3 | P97429 | Anxa4 | Annexin A4 | 2 | 2.2109 |
| 6.95 | 86.7 | P11591 | Mup5 | Major urinary protein 5 | 50 | 2.2037 |
| 6.09 | 23.7 | Q60590 | Orm1 | Alpha-1-acid glycoprotein 1 | 3 | 2.2033 |
| 4.02 | 34 | P01837 | Igkc | Ig kappa chain C region | 2 | 2.1829 |
| 2.25 | 5.4 | P97434-3 | Mprip | Isoform 3 of Myosin phosphatase Rho-interacting protein | 1 | 2.1059 |
| 2.28 | 10.8 | Q61147 | Cp | Ceruloplasmin | 1 | 2.0868 |
| 8.05 | 18.9 | Q07456 | Ambp | Protein AMBP | 4 | 2.084 |
| 12.89 | 44.4 | O09114 | Ptgds | Prostaglandin-H2 D-isomerase | 7 | 1.9728 |
| 45.67 | 44.7 | P07724 | Alb | Serum albumin | 23 | 1.8624 |
| 105.92 | 51.3 | P28825 | Mep1a | Meprin A subunit alpha | 96 | 1.8616 |
| 2.27 | 15.2 | Q9JI02 | Scgb2b20 | Secretoglobin family 2B member 20 | 1 | 1.8537 |
| 4 | 7.8 | Q01279 | Egfr | Epidermal growth factor receptor | 2 | 1.852 |
| 8.13 | 35.7 | P15501 | Sbp | Prostatic spermine-binding protein | 5 | 1.8465 |
| 113.02 | 84.5 | P00688 | Amy2 | Pancreatic alpha-amylase | 97 | 1.8379 |
| 3.07 | 5.4 | Q07797 | Lgals3bp | Galectin-3-binding protein | 1 | 1.7984 |
| 20.01 | 32.1 | P07759 | Serpina3k | Serine protease inhibitor A3K | 14 | 1.7874 |
| 10.57 | 81.8 | Q5FW60 | Mup20 | Major urinary protein 20 | 104 | 1.7659 |
| 3.75 | 18.1 | P46412 | Gpx3 | Glutathione peroxidase 3 | 2 | 1.7549 |
| 138.4 | 87.5 | P04939 | Mup3 | Major urinary protein 3 | 218 | 1.7381 |
| 46.37 | 65.6 | Q00897 | Serpina1d | Alpha-1-antitrypsin 1-4 | 37 | 1.7123 |
| 14.42 | 37.7 | Q8C6C9 | Leg1 | Protein LEG1 homolog | 10 | 1.7123 |
| 2.07 | 8.1 | Q9CZ52-2 | Antxr1 | Isoform 2 of Anthrax toxin receptor 1 | 1 | 1.7001 |
| 2.01 | 12 | O35298 | Aoah | Acyloxyacyl hydrolase | 1 | 1.697 |
| 24.53 | 52.5 | P15947 | Klk1 | Kallikrein-1 | 23 | 1.6927 |
| 10.38 | 33 | P13634 | Ca1 | Carbonic anhydrase 1 | 6 | 1.6857 |
| 2.57 | 16.6 | P06869 | Plau | Urokinase-type plasminogen activator | 1 | 1.6756 |
| 5.67 | 58.4 | Q00898 | Serpina1e | Alpha-1-antitrypsin 1-5 | 29 | 1.661 |
| 11.55 | 64.4 | P07758 | Serpina1a | Alpha-1-antitrypsin 1-1 | 36 | 1.6561 |
| 5.13 | 11.4 | P01898 | H2-Q10 | H-2 class I histocompatibility antigen, Q10 alpha chain | 2 | 1.6519 |
| 19.02 | 29 | Q9R1E6 | Enpp2 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 | 11 | 1.6432 |
| 4.1 | 9.1 | Q61391 | Mme | Neprilysin | 2 | 1.629 |
| 4.01 | 12.6 | Q99LJ1 | Fuca1 | Tissue alpha-L-fucosidase | 2 | 1.6238 |
| 6.05 | 29.2 | P10605 | Ctsb | Cathepsin B | 3 | 1.6108 |
| 2.02 | 7.9 | P04925 | Prnp | Major prion protein | 1 | 1.5938 |
| 4.17 | 10.4 | Q571E4 | Galns | N-acetylgalactosamine-6-sulfatase | 2 | 1.5885 |
| 18.01 | 42.1 | P03953 | Cfd | Complement factor D | 12 | 1.5799 |
| 6.15 | 28.8 | P02816 | Pip | Prolactin-inducible protein homolog | 3 | 1.5788 |
| 10.89 | 11.5 | P97449 | Anpep | Aminopeptidase N | 6 | 1.562 |
| 19.03 | 20.7 | P23780 | Glb1 | Beta-galactosidase | 10 | 1.5572 |
| 3.52 | 11.9 | Q61592 | Gas6 | Growth arrest-specific protein 6 | 2 | 1.5507 |
| 6 | 10.6 | P70269 | Ctse | Cathepsin E | 4 | 1.5426 |
| 22.14 | 20.2 | P09470 | Ace | Angiotensin-converting enzyme | 11 | 1.5142 |
| 4.02 | 16.5 | Q9DBV4 | Mxra8 | Matrix remodeling-associated protein 8 | 2 | 1.5131 |
| 2.02 | 7.9 | P23953 | Ces1c | Carboxylesterase 1C | 1 | 1.5105 |
| 30.67 | 50 | P49183 | Dnase1 | Deoxyribonuclease-1 | 17 | 1.4851 |
| 4 | 9 | P20060 | Hexb | Beta-hexosaminidase subunit beta | 2 | 1.4841 |
| 4.29 | 86.7 | P11588 | Mup1 | Major urinary protein 1 | 331 | 1.4812 |
| 6.03 | 13 | Q61398 | Pcolce | Procollagen C-endopeptidase enhancer 1 | 3 | 1.469 |
| 3.7 | 16.4 | Q60963 | Pla2g7 | Platelet-activating factor acetylhydrolase | 2 | 1.4604 |
| 8.6 | 13.7 | P16675 | Ctsa | Lysosomal protective protein | 4 | 1.4516 |
| 4.2 | 15.2 | Q9Z0K8 | Vnn1 | Pantetheinase | 4 | 1.447 |
| 4 | 15.9 | P63101 | Ywhaz | 14-3-3 protein zeta/delta | 2 | 1.4451 |
| 7.78 | 17.1 | Q6GU68 | Islr | Immunoglobulin superfamily containing leucine-rich repeat protein | 4 | 1.4442 |
| 4.06 | 9.4 | P27046 | Man2a1 | Alpha-mannosidase 2 | 2 | 1.4354 |
| 16 | 21.3 | Q61847 | Mep1b | Meprin A subunit beta | 9 | 1.4333 |
| 5.23 | 16.5 | Q9WV54 | Asah1 | Acid ceramidase | 2 | 1.4315 |
| 4.69 | 13.1 | C0HKG6 | Rnaset2b | Ribonuclease T2-B | 3 | 1.41 |
| 4.05 | 5.7 | O54782 | Man2b2 | Epididymis-specific alpha-mannosidase | 2 | 1.4055 |
| 4 | 22.5 | P19 639 | Gstm3 | Glutathione S-transferase Mu 3 | 2 | 1.3973 |
| 5.3 | 6.1 | Q8R0I0 | Ace2 | Angiotensin-converting enzyme 2 | 3 | 1.3865 |
| 15.4 | 30.7 | P63260 | Actg1 | Actin, cytoplasmic 2 | 10 | 1.373 |
| 2.12 | 10.4 | P24638 | Acp2 | Lysosomal acid phosphatase | 1 | 1.3441 |
| 9.59 | 5.7 | Q5SSE9 | Abca13 | ATP-binding cassette sub-family A member 13 | 4 | 1.3385 |
| 5.7 | 35.9 | P62806 | Hist1h4a | Histone H4 | 3 | 1.3277 |
| 5.07 | 12.3 | Q9QWR8 | Naga | Alpha-N-acetylgalactosaminidase | 2 | 1.3186 |
| 17.26 | 16.5 | O09159 | Man2b1 | Lysosomal alpha-mannosidase | 8 | 1.3091 |
| 2.03 | 6.5 | Q9WUU7 | Ctsz | Cathepsin Z | 1 | 1.2977 |
| 3.94 | 14.7 | O35082 | Kl | Klotho | 1 | 1.2901 |
| 2.02 | 8.1 | Q9ESG4 | Tmem27 | Collectrin | 1 | 1.2855 |
| 12.08 | 29.1 | Q9JIL4 | Pdzk1 | Na(+)/H(+) exchange regulatory cofactor NHE-RF3 | 6 | 1.2789 |
| 14.34 | 17.7 | Q91WV7 | Slc3a1 | Neutral and basic amino acid transport protein rBAT | 7 | 1.2712 |
| 4 | 11.7 | Q9CYN9 | Atp6ap2 | Renin receptor | 2 | 1.2534 |
| 10.16 | 43.2 | Q9CQ60 | Pgls | 6-phosphogluconolactonase | 5 | 1.2505 |
| 6 | 12.9 | P49935 | Ctsh | Pro-cathepsin H | 3 | 1.2393 |
| 2.08 | 7.7 | Q8K209 | Adgrg1 | Adhesion G-protein coupled receptor G1 | 1 | 1.2388 |
| 4 | 12.4 | Q03404 | Tff2 | Trefoil factor 2 | 2 | 1.2351 |
| 4.06 | 19.2 | O89051 | Itm2b | Integral membrane protein 2B | 2 | 1.2343 |
| 8.13 | 26.7 | P08226 | Apoe | Apolipoprotein E | 5 | 1.2285 |
| 5.7 | 10.4 | Q9CZT5 | Vasn | Vasorin | 3 | 1.2261 |
| 16.33 | 10.9 | Q61838 | Pzp | Pregnancy zone protein | 8 | 1.226 |
| 63.56 | 61.2 | Q06890 | Clu | Clusterin | 50 | 1.2243 |
| 23.36 | 25.4 | P70699 | Gaa | Lysosomal alpha-glucosidase | 16 | 1.2238 |
| 12.12 | 22.1 | O89023 | Tpp1 | Tripeptidyl-peptidase 1 | 6 | 1.2174 |
| 3.3 | 16.1 | P63082 | Atp6v0c | V-type proton ATPase 16 kDa proteolipid subunit | 3 | 1.2157 |
| Gene Symbol | UniProt Accession | Name | U-exo | N-exo |
|---|---|---|---|---|
| Zfp292 | Q9Z2U2 | Zinc finger protein 292 | √ | √ |
| Spen | Q62504-3 | Isoform 3 of Msx2-interacting protein | × | √ |
| Smg1 | Q8BKX6 | Serine/threonine-protein kinase SMG1 | √ | √ |
| Ryr2 | E9Q401 | Ryanodine receptor 2 | √ | √ |
| Ptgds | O09114 | Prostaglandin-H2 D-isomerase | √ | √ |
| Myt1l | P97500 | Myelin transcription factor 1-like protein | × | √ |
| Myo9a | Q8C170 | Unconventional myosin-IXa | √ | √ |
| Morc2a | Q69ZX6 | MORC family CW-type zinc finger protein 2A | × | √ |
| Kif21b | Q9QXL1 | Kinesin-like protein KIF21B | √ | √ |
| Kdm2b | Q6P1G2 | Lysine-specific demethylase 2B | × | √ |
| Kcna4 | Q61423 | Potassium voltage-gated channel subfamily A member 4 | × | √ |
| Igkc | P01837 | Ig kappa chain C region | × | √ |
| Epc2 | Q8C0I4 | Enhancer of polycomb homolog 2 | × | √ |
| Cacna1b | O55017 | Voltage-dependent N-type calcium channel subunit alpha-1B | × | √ |
| Atp1b1 | P14094 | Sodium/potassium-transporting ATPase subunit beta-1 | √ | √ |
| BP Terms | U-exo | N-exo | ||
|---|---|---|---|---|
| Gene Symbol | Fold Change | Gene Symbol | Fold Change | |
| Sphingolipid catabolic process | Asah1 | 1.3824 | Asah1 | 1.4315 |
| Galc | 1.1177 | Galc | 0.9068 | |
| Hexb | 1.1909 | Hexb | 1.4841 | |
| Smpdl3a | 1.3852 | Smpdl3a | 1.1356 | |
| Smpd1 | 1.3335 | Naga | 1.3186 | |
| Enpp2 | 1.6432 | |||
| Ceramide catabolic process | Asah1 | 1.3824 | Asah1 | 1.4315 |
| Galc | 1.1177 | Galc | 0.9068 | |
| Hexb | 1.1909 | Hexb | 1.4841 | |
| Naga | 1.3186 | |||
| Membrane lipid catabolic process | Asah1 | 1.3824 | Asah1 | 1.4315 |
| Galc | 1.1177 | Galc | 0.9068 | |
| Hexb | 1.1909 | Hexb | 1.4841 | |
| Smpdl3a | 1.3852 | Smpdl3a | 1.1356 | |
| Smpd1 | 1.3335 | Naga | 1.3186 | |
| Enpp2 | 1.6432 | |||
| Aβ clearance | Apoe | 1.4288 | Apoe | 1.2285 |
| Clu | 1.3055 | Clu | 1.2243 | |
| Lrp2 | 1.1960 | Lrp2 | 1.16 | |
| Mme | 1.6804 | Mme | 1.629 | |
| Aβ metabolic process | Ace | 1.5564 | Ace | 1.5142 |
| Apoe | 1.4288 | Apoe | 1.2285 | |
| Clu | 1.3055 | Clu | 1.2243 | |
| Mme | 1.6804 | Mme | 1.629 | |
| Prnp | 1.5938 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Sheikh, A.M.; Matsumoto, K.-i.; Mitaki, S.; Shibly, A.Z.; Zhang, Y.; A, G.; Yano, S.; Nagai, A. iTRAQ-Based Proteomic Analysis of APP Transgenic Mouse Urine Exosomes. Int. J. Mol. Sci. 2023, 24, 672. https://doi.org/10.3390/ijms24010672
Zhou X, Sheikh AM, Matsumoto K-i, Mitaki S, Shibly AZ, Zhang Y, A G, Yano S, Nagai A. iTRAQ-Based Proteomic Analysis of APP Transgenic Mouse Urine Exosomes. International Journal of Molecular Sciences. 2023; 24(1):672. https://doi.org/10.3390/ijms24010672
Chicago/Turabian StyleZhou, Xiaojing, Abdullah Md. Sheikh, Ken-ichi Matsumoto, Shingo Mitaki, Abu Zaffar Shibly, Yuchi Zhang, Garu A, Shozo Yano, and Atsushi Nagai. 2023. "iTRAQ-Based Proteomic Analysis of APP Transgenic Mouse Urine Exosomes" International Journal of Molecular Sciences 24, no. 1: 672. https://doi.org/10.3390/ijms24010672
APA StyleZhou, X., Sheikh, A. M., Matsumoto, K. -i., Mitaki, S., Shibly, A. Z., Zhang, Y., A, G., Yano, S., & Nagai, A. (2023). iTRAQ-Based Proteomic Analysis of APP Transgenic Mouse Urine Exosomes. International Journal of Molecular Sciences, 24(1), 672. https://doi.org/10.3390/ijms24010672