
Chinese Pedigree with Hereditary Gastrointestinal Stromal Tumors: A Case Report and Literature Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
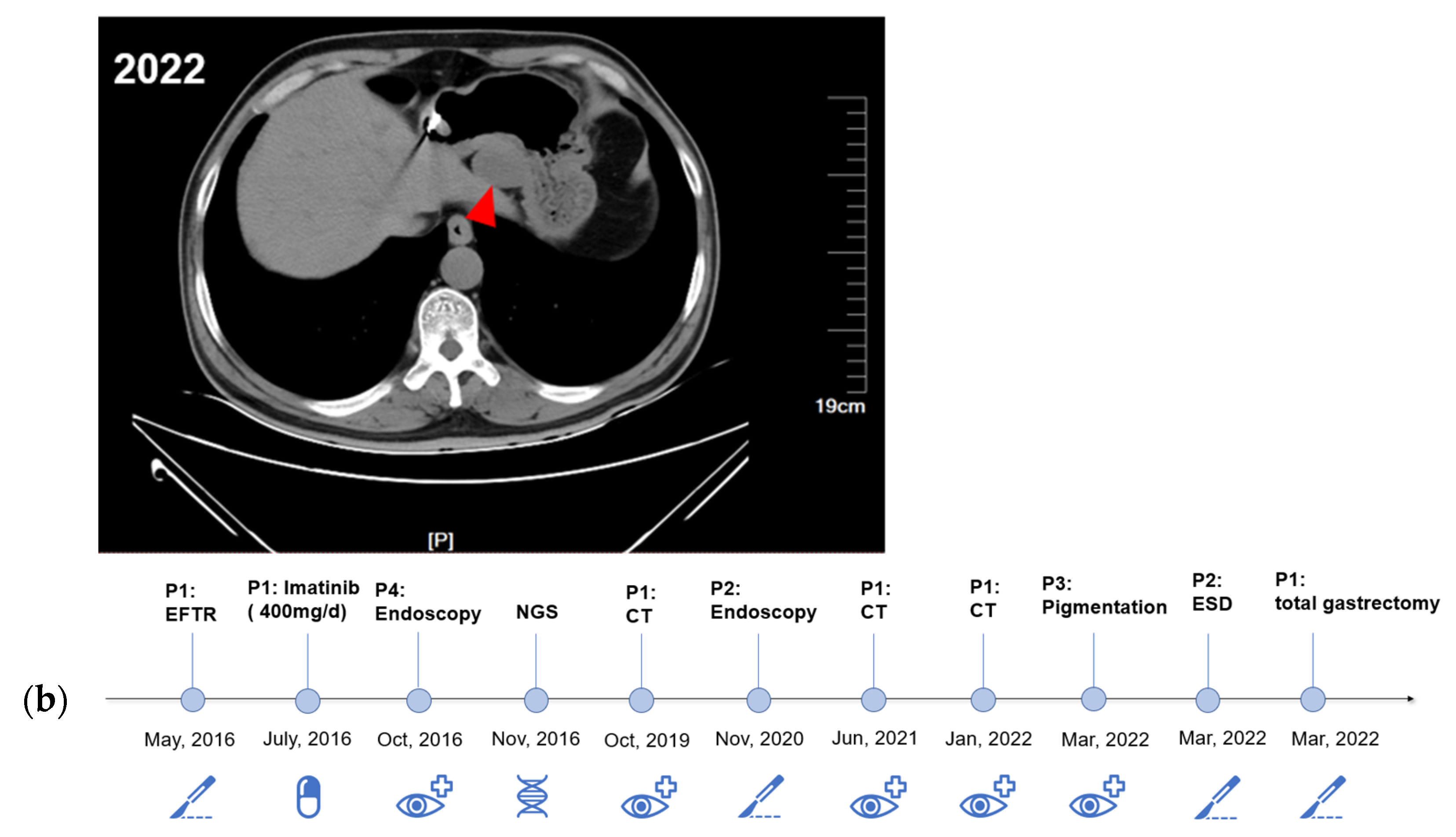
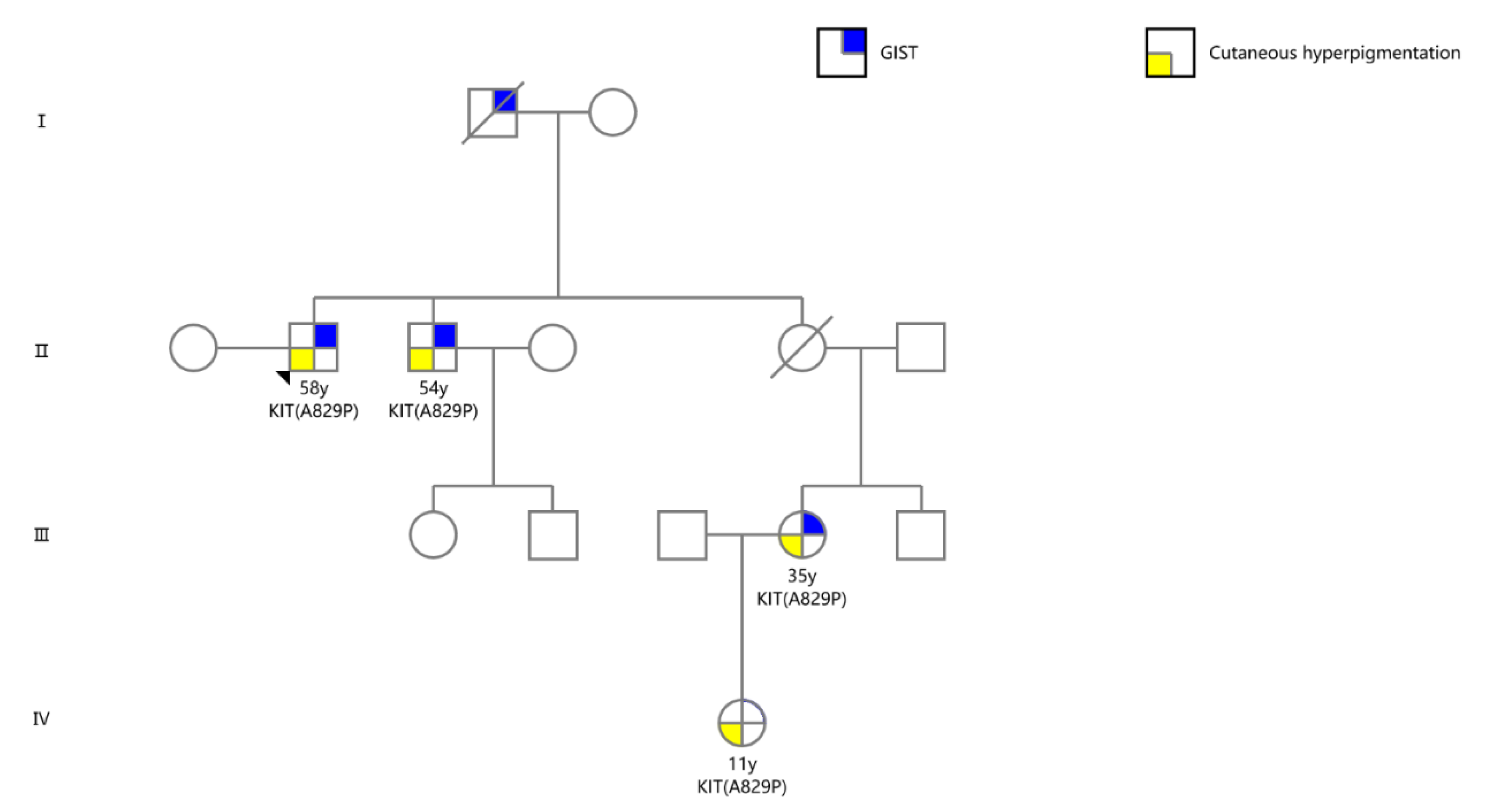
2.1. Case Presentation
2.2. DNA Analyses
2.3. Candidate Targeted Drugs Screening
3. Discussion
4. Conclusions
5. Methods and Materials
5.1. Clinical Diagnosis and Treatment
5.2. Molecular Analysis
5.3. Cell Culture, Mutant Construction, Plasmid Transfection, and Cell Viability Assay
5.4. Western Blotting
5.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Poveda, A.; García del Muro, X.; López-Guerrero, J.A.; Cubedo, R.; Martínez, V.; Romero, I.; Serrano, C.; Valverde, C.; Martín-Broto, J. GEIS Guidelines for Gastrointestinal Sarcomas (GIST). Cancer Treat. Rev. 2017, 55, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, C.M.; Gutierrez Sainz, L.; Chi, P. The Management of Metastatic GIST: Current Standard and Investigational Therapeutics. J. Hematol. Oncol. 2021, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; George, S. Gastrointestinal Stromal Tumor: Challenges and Opportunities for a New Decade. Clin. Cancer Res. 2020, 26, 5078–5085. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.-Y.; Kang, Y.-K.; Nishida, T.; von Mehren, M. Gastrointestinal Stromal Tumours. Nat. Rev. Dis. Prim. 2021, 7, 22. [Google Scholar] [CrossRef]
- Arima, J.; Hiramatsu, M.; Taniguchi, K.; Kobayashi, T.; Tsunematsu, I.; Kagota, S.; Sakane, J.; Suzuki, Y.; Hirota, S. Multiple Gastrointestinal Stromal Tumors Caused by a Novel Germline KIT Gene Mutation (Asp820Gly): A Case Report and Literature Review. Gastric Cancer 2020, 23, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Fornasarig, M.; Gasparotto, D.; Foltran, L.; Campigotto, M.; Lombardi, S.; Del Savio, E.; Buonadonna, A.; Puglisi, F.; Sulfaro, S.; Canzonieri, V.; et al. A Novel Kindred with Familial Gastrointestinal Stromal Tumors Caused by a Rare KIT Germline Mutation (N655K): Clinico-Pathological Presentation and TKI Sensitivity. J. Pers. Med. 2020, 10, 234. [Google Scholar] [CrossRef]
- Wali, G.N.; Halliday, D.; Dua, J.; Ieremia, E.; McPherson, T.; Matin, R.N. Cutaneous Hyperpigmentation and Familial Gastrointestinal Stromal Tumour Associated with KIT Mutation. Clin. Exp. Dermatol. 2019, 44, 418–421. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, D.; Liu, Y.; Chen, W.; Liu, M.; Xie, C.; Wang, J. Multiple Gastrointestinal Stromal Tumors with Exon 11 Mutation of the C-KIT Gene in a Young Male without Family History. Int. J. Clin. Exp. Pathol. 2020, 13, 1766–1770. [Google Scholar]
- Neuhann, T.M.; Mansmann, V.; Merkelbach-Bruse, S.; Klink, B.; Hellinger, A.; Höffkes, H.-G.; Wardelmann, E.; Schildhaus, H.-U.; Tinschert, S. A Novel Germline KIT Mutation (p.L576P) in a Family Presenting with Juvenile Onset of Multiple Gastrointestinal Stromal Tumors, Skin Hyperpigmentations, and Esophageal Stenosis. Am. J. Surg. Pathol. 2013, 37, 898–905. [Google Scholar] [CrossRef]
- Ke, H.; Kazi, J.U.; Zhao, H.; Sun, J. Germline Mutations of KIT in Gastrointestinal Stromal Tumor (GIST) and Mastocytosis. Cell Biosci. 2016, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Tran, A.K.; Pearce, A.; López-Sánchez, M.; Pérez-Jurado, L.A.; Barnett, C. Novel KIT Mutation Presenting as Marked Lentiginosis. Pediatr. Dermatol. 2019, 36, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Sugiura, K.; Tanahashi, K.; Noda, K.; Kono, M.; Akiyama, M. Autosomal Dominant Progressive Hyperpigmentation and Lentigines in a Japanese Pedigree Due to a Missense Mutation near the C-Terminus of KIT. Br. J. Dermatol. 2018, 179, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Y.; Wang, T. Case Report: A Missense Mutation of KIT in Hyperpigmentation and Lentigines Unassociated With Systemic Disorders: Report of a Chinese Pedigree and a Literature Review. Front. Med. 2022, 9, 847382. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Sofuni, A.; Tsuchiya, T.; Kono, S.; Ishii, K.; Tanaka, R.; Tonozuka, R.; Mukai, S.; Yamamoto, K.; Matsunami, Y.; et al. Efficacy of the Franseen Needle for Diagnosing Gastrointestinal Submucosal Lesions Including Small Tumors. Endosc. Ultrasound 2021, 10, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Kuwatani, M.; Sakamoto, N. Evolution and a Promising Role of EUS-FNA in Gene and Future Analyses. Endosc. Ultrasound 2020, 9, 151–153. [Google Scholar] [CrossRef]
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Ge, Q.; Yang, F.; Wang, S.; Ge, N.; Liu, X.; Shi, J.; Fusaroli, P.; Liu, Y.; Sun, S. Small Gastric Stromal Tumors: An Underestimated Risk. Cancers 2022, 14, 6008. [Google Scholar] [CrossRef]
- Maeyama, H.; Hidaka, E.; Ota, H.; Minami, S.; Kajiyama, M.; Kuraishi, A.; Mori, H.; Matsuda, Y.; Wada, S.; Sodeyama, H.; et al. Familial Gastrointestinal Stromal Tumor with Hyperpigmentation: Association with a Germline Mutation of the c-Kit Gene. Gastroenterology 2001, 120, 210–215. [Google Scholar] [CrossRef]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial Gastrointestinal Stromal Tumours with Germline Mutation of the KIT Gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Todd, J.R.; Becker, T.M.; Kefford, R.F.; Rizos, H. Secondary C-Kit Mutations Confer Acquired Resistance to RTK Inhibitors in c-Kit Mutant Melanoma Cells. Pigment Cell Melanoma Res. 2013, 26, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.-A.; Jacques, S.L.; et al. KIT Kinase Mutants Show Unique Mechanisms of Drug Resistance to Imatinib and Sunitinib in Gastrointestinal Stromal Tumor Patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-K.; Wu, C.-E.; Wang, S.-Y.; Chang, C.-F.; Chou, W.-C.; Chen, J.-S.; Yeh, C.-N. Systemic Therapy for Gastrointestinal Stromal Tumor: Current Standards and Emerging Challenges. Curr. Treat. Options Oncol. 2022, 23, 1303–1319. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and Safety of Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumour after Failure of Imatinib: A Randomised Controlled Trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- George, S.; Wang, Q.; Heinrich, M.C.; Corless, C.L.; Zhu, M.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Tap, W.D.; et al. Efficacy and Safety of Regorafenib in Patients With Metastatic and/or Unresectable GI Stromal Tumor After Failure of Imatinib and Sunitinib: A Multicenter Phase II Trial. JCO 2012, 30, 2401–2407. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in Patients with Advanced Gastrointestinal Stromal Tumours (INVICTUS): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Bauer, S.; Blay, J.-Y.; Choucair, K.; Gelderblom, H.; George, S.; Schöffski, P.; von Mehren, M.; Zalcberg, J.; Achour, H.; et al. Intrigue: Phase III Study of Ripretinib versus Sunitinib in Advanced Gastrointestinal Stromal Tumor after Imatinib. Future Oncol. 2020, 16, 4251–4264. [Google Scholar] [CrossRef] [Green Version]
- Goggin, C.; Stansfeld, A.; Mahalingam, P.; Thway, K.; Smith, M.J.; Huang, P.; Jones, R.L.; Napolitano, A. Ripretinib in Advanced Gastrointestinal Stromal Tumors: An Overview of Current Evidence and Drug Approval. Future Oncol. 2022, 18, 2967–2978. [Google Scholar] [CrossRef]
- Dhillon, S. Ripretinib: First Approval. Drugs 2020, 80, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.; Gardino, A.; Hodous, B.; Davis, A.; Zhu, J.; Kohl, N.E.; Lengauer, C. Blu-285, a Potent and Selective Inhibitor for Hematologic Malignancies with KIT Exon 17 Mutations. Blood 2015, 126, 568. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, Q.; Liu, Y.; Yang, F.; Sun, G.; Guo, J.; Sun, S. Chinese Pedigree with Hereditary Gastrointestinal Stromal Tumors: A Case Report and Literature Review. Int. J. Mol. Sci. 2023, 24, 830. https://doi.org/10.3390/ijms24010830
Ge Q, Liu Y, Yang F, Sun G, Guo J, Sun S. Chinese Pedigree with Hereditary Gastrointestinal Stromal Tumors: A Case Report and Literature Review. International Journal of Molecular Sciences. 2023; 24(1):830. https://doi.org/10.3390/ijms24010830
Chicago/Turabian StyleGe, Qichao, Yang Liu, Fan Yang, Guangwei Sun, Jintao Guo, and Siyu Sun. 2023. "Chinese Pedigree with Hereditary Gastrointestinal Stromal Tumors: A Case Report and Literature Review" International Journal of Molecular Sciences 24, no. 1: 830. https://doi.org/10.3390/ijms24010830
APA StyleGe, Q., Liu, Y., Yang, F., Sun, G., Guo, J., & Sun, S. (2023). Chinese Pedigree with Hereditary Gastrointestinal Stromal Tumors: A Case Report and Literature Review. International Journal of Molecular Sciences, 24(1), 830. https://doi.org/10.3390/ijms24010830