
Multiplexed Gene Engineering Based on dCas9 and gRNA-tRNA Array Encoded on Single Transcript

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
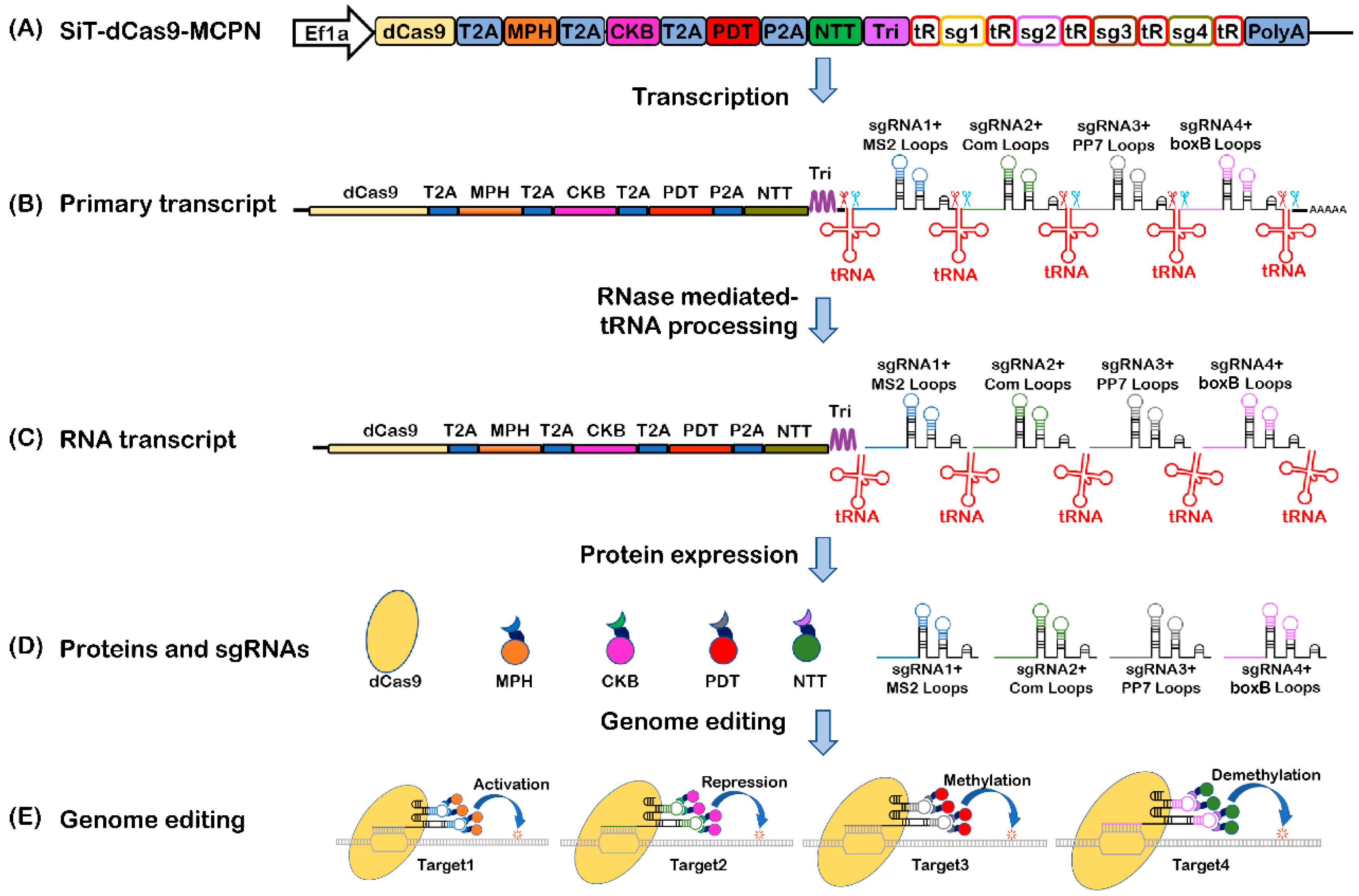
2.1. Construction and Mechanism of the Multifunctional System Based on CRISPR/dCas9 and tRNA-gRNA Array
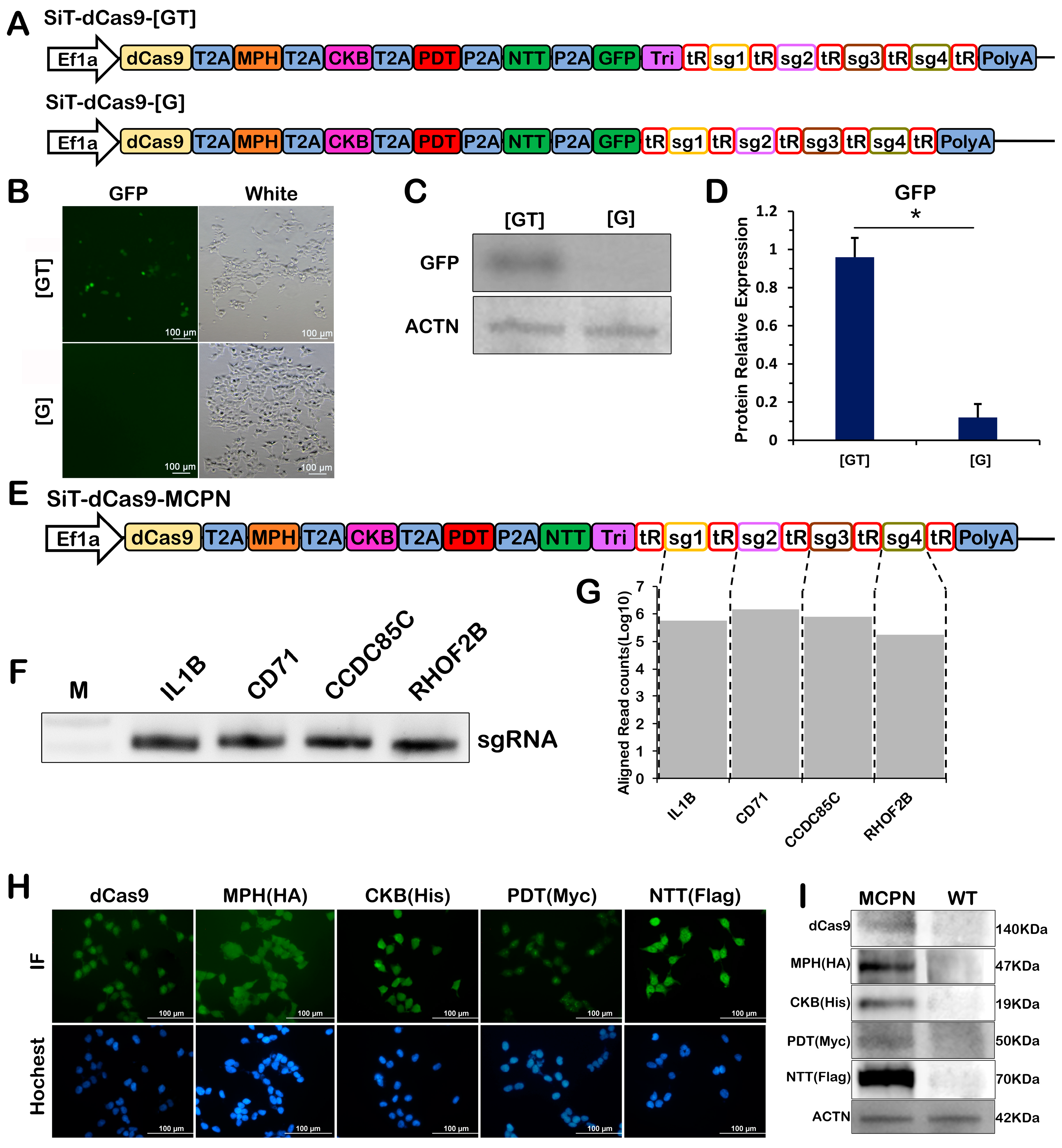
2.2. Simultaneous Expression of Multiple Proteins and gRNA Scaffolds from a Single Pol II-Derived Transcript
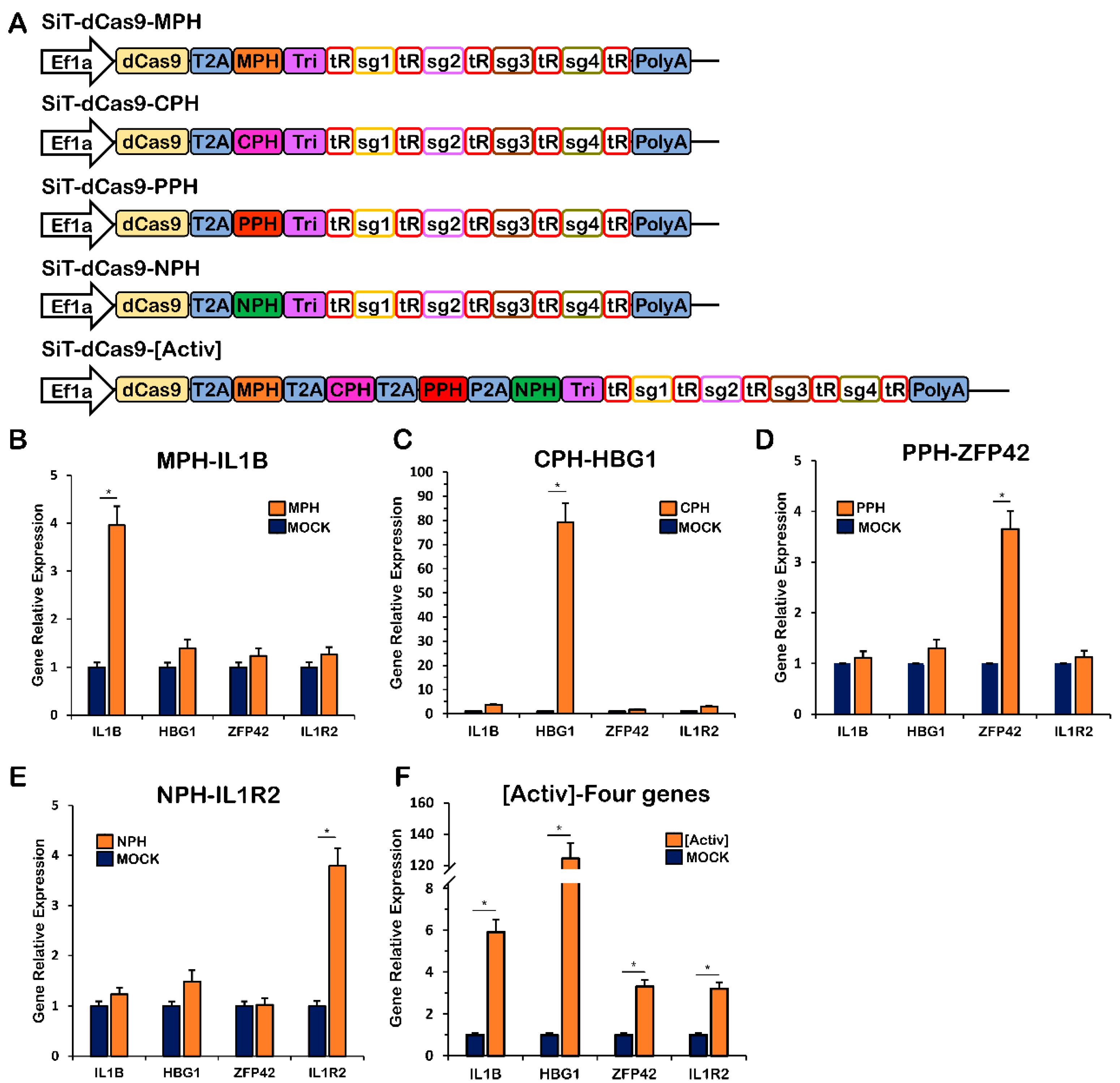
2.3. Simultaneous Gene Activation, Gene Repression, DNA Methylation and DNA Demethylation with the CRISPR-MCPN System
2.4. Specific Interaction of the MS2, PP7, Com or λ N22 RNA Stem–Loop with Its RNA-Binding Protein
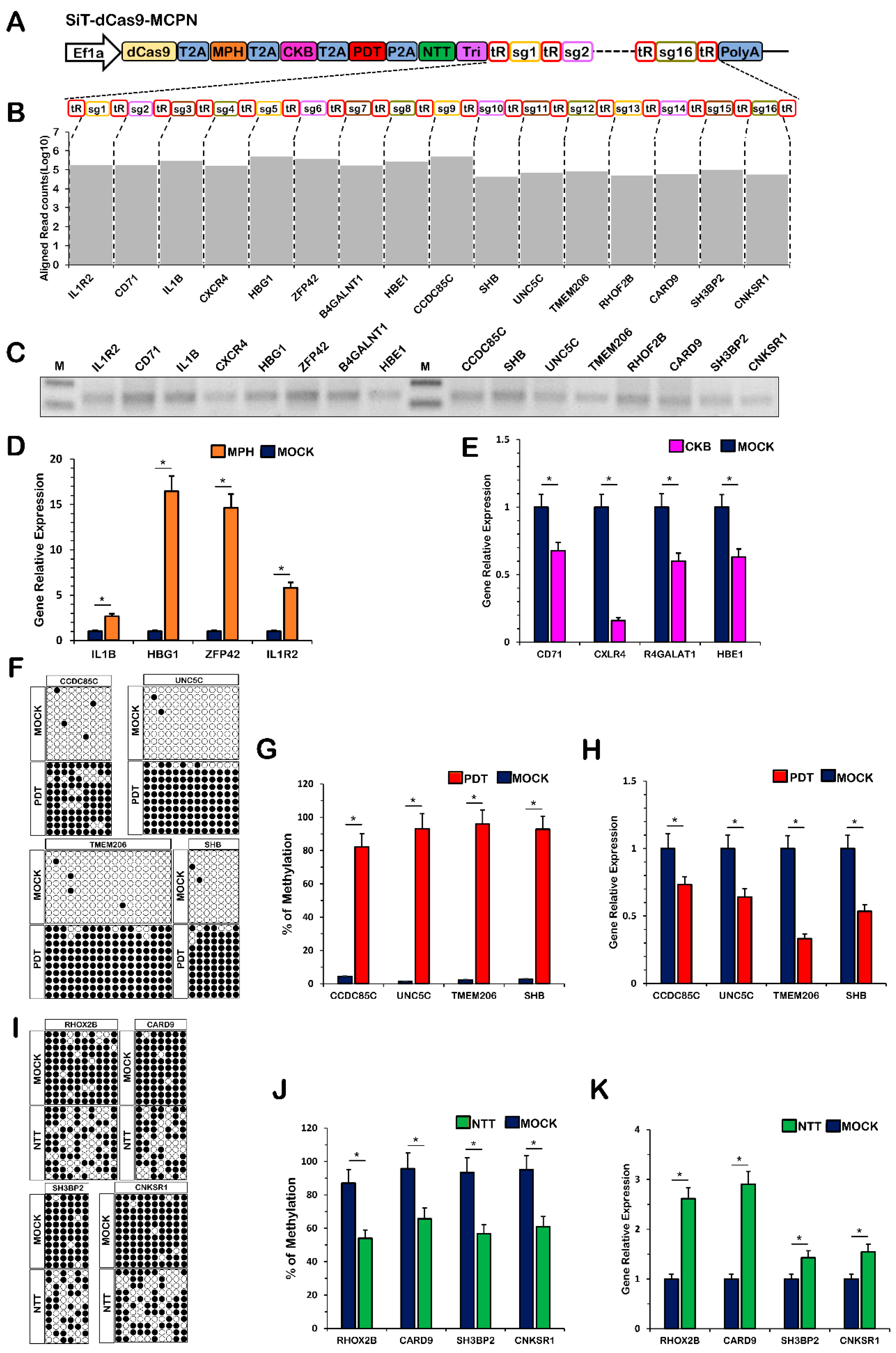
2.5. Simultaneous Multiplexed Genome Editing with the CRISPR-MCPN System
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Transfection
4.3. Plasmid Construction
4.4. Quantification of mRNA Expression
4.5. Western Blot
4.6. Immunocytochemistry
4.7. Mature gRNA Quantification
4.8. Targeted BS-PCR Primer Design and Sequencing
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, W.; Zhang, S.; Liu, C.C.; Zhou, X.J. Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics 2012, 28, 2458–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Zhang, Y.; Weng, W.; Chen, J.; Cai, H. Survey and comparative assessments of computational multi-omics integrative methods with multiple regulatory networks identifying distinct tumor compositions across pan-cancer data sets. Brief. Bioinform. 2021, 22, bbaa102. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Gao, L.; Gao, Y.; Hu, Y.; Xu, H.; Huang, M.; Song, K.; Wang, H.; Dong, Y.; Jiang, C.; et al. Evaluation and comparison of multi-omics data integration methods for cancer subtyping. PLoS Comput. Biol. 2021, 17, e1009224. [Google Scholar] [CrossRef] [PubMed]
- Drouard, G.; Ollikainen, M.; Mykkanen, J.; Raitakari, O.; Lehtimaki, T.; Kahonen, M.; Mishra, P.P.; Wang, X.; Kaprio, J. Multi-Omics Integration in a Twin Cohort and Predictive Modeling of Blood Pressure Values. OMICS 2022, 26, 130–141. [Google Scholar] [CrossRef]
- Eddy, S.; Mariani, L.H.; Kretzler, M. Integrated multi-omics approaches to improve classification of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lin, Q.; Jin, S.; Gao, C. The CRISPR-Cas toolbox and gene editing technologies. Mol. Cell 2022, 82, 333–347. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Nunez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Zhang, W.; Hu, H.; Xue, B.; Qin, J.; Sun, C.; Sun, Y.; Wei, W.; Sun, Y. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res. 2016, 44, e86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. [Google Scholar] [CrossRef] [Green Version]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zong, Y.; Jin, S.; Zhu, H.; Lin, D.; Li, S.; Qiu, J.-L.; Wang, Y.; Gao, C. SWISS: Multiplexed orthogonal genome editing in plants with a Cas9 nickase and engineered CRISPR RNA scaffolds. Genome Biol. 2020, 21, 141. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Skrekas, C.; Nielsen, J.; David, F. Multiplexed CRISPR/Cas9 Genome Editing and Gene Regulation Using Csy4 in Saccharomyces cerevisiae. ACS Synth. Biol. 2018, 7, 10–15. [Google Scholar] [CrossRef]
- Yuan, Q.; Gao, X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat. Commun. 2022, 13, 2771. [Google Scholar] [CrossRef]
- He, X.; Wang, Y.; Yang, F.; Wang, B.; Xie, H.; Gu, L.; Zhao, T.; Liu, X.; Zhang, D.; Ren, Q.; et al. Boosting activity of high-fidelity CRISPR/Cas9 variants using a tRNA(Gln)-processing system in human cells. J. Biol. Chem. 2019, 294, 9308–9315. [Google Scholar] [CrossRef]
- Abdelrahman, M.; Wei, Z.; Rohila, J.S.; Zhao, K. Multiplex Genome-Editing Technologies for Revolutionizing Plant Biology and Crop Improvement. Front. Plant. Sci. 2021, 12, 721203. [Google Scholar] [CrossRef]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [Green Version]
- Nissim, L.; Perli, S.D.; Fridkin, A.; Perez-Pinera, P.; Lu, T.K. Multiplexed and Programmable Regulation of Gene Networks with an Integrated RNA and CRISPR/Cas Toolkit in Human Cells. Mol. Cell 2014, 54, 698–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlman, J.E.; Abudayyeh, O.O.; Joung, J.; Gootenberg, J.S.; Zhang, F.; Konermann, S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015, 33, 1159–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.; HamediRad, M.; Hu, S.; Zhao, H. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat. Commun. 2017, 8, 1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7, 11707. [Google Scholar] [CrossRef] [Green Version]
- Haurwitz, R.E.; Jinek, M.; Wiedenheft, B.; Zhou, K.; Doudna, J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 2010, 329, 1355–1358. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Chen, K.; Chen, Y.; Li, H.; Xie, K. Engineering Introns to Express RNA Guides for Cas9- and Cpf1-Mediated Multiplex Genome Editing. Mol. Plant 2018, 11, 542–552. [Google Scholar] [CrossRef] [Green Version]
- McCarty, N.S.; Graham, A.E.; Studena, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Nistico, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res. Rev. 2021, 69, 101346. [Google Scholar] [CrossRef] [PubMed]
- Abyadeh, M.; Tofigh, N.; Hosseinian, S.; Hasan, M.; Amirkhani, A.; Fitzhenry, M.J.; Gupta, V.; Chitranshi, N.; Salekdeh, G.H.; Haynes, P.A.; et al. Key Genes and Biochemical Networks in Various Brain Regions Affected in Alzheimer’s Disease. Cells 2022, 11, 987. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Geng, L.; Wang, J.; Liang, Y.; Guo, X.; Liu, C.; Zhao, Y.; Jin, J.; Liu, Z.; Mu, Y. Multiplexed Gene Engineering Based on dCas9 and gRNA-tRNA Array Encoded on Single Transcript. Int. J. Mol. Sci. 2023, 24, 8535. https://doi.org/10.3390/ijms24108535
Jiang C, Geng L, Wang J, Liang Y, Guo X, Liu C, Zhao Y, Jin J, Liu Z, Mu Y. Multiplexed Gene Engineering Based on dCas9 and gRNA-tRNA Array Encoded on Single Transcript. International Journal of Molecular Sciences. 2023; 24(10):8535. https://doi.org/10.3390/ijms24108535
Chicago/Turabian StyleJiang, Chaoqian, Lishuang Geng, Jinpeng Wang, Yingjuan Liang, Xiaochen Guo, Chang Liu, Yunjing Zhao, Junxue Jin, Zhonghua Liu, and Yanshuang Mu. 2023. "Multiplexed Gene Engineering Based on dCas9 and gRNA-tRNA Array Encoded on Single Transcript" International Journal of Molecular Sciences 24, no. 10: 8535. https://doi.org/10.3390/ijms24108535
APA StyleJiang, C., Geng, L., Wang, J., Liang, Y., Guo, X., Liu, C., Zhao, Y., Jin, J., Liu, Z., & Mu, Y. (2023). Multiplexed Gene Engineering Based on dCas9 and gRNA-tRNA Array Encoded on Single Transcript. International Journal of Molecular Sciences, 24(10), 8535. https://doi.org/10.3390/ijms24108535