

Mitochondrial Trafficking of MLKL, Bak/Bax, and Drp1 Is Mediated by RIP1 and ROS which Leads to Decreased Mitochondrial Membrane Integrity during the Hyperglycemic Shift to Necroptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
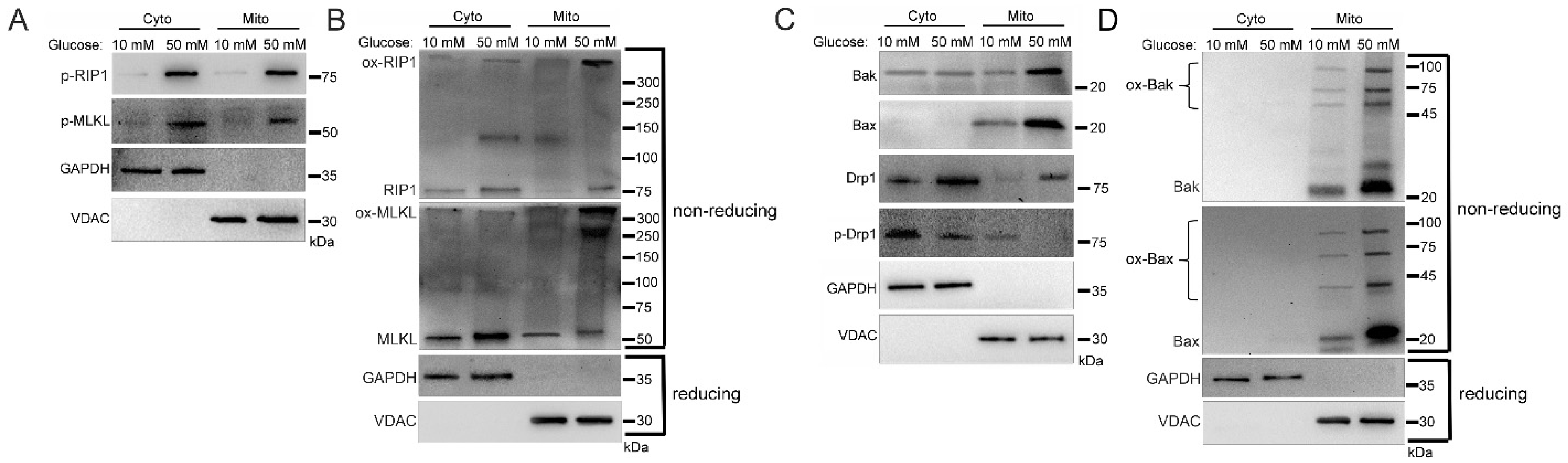
2.1. Increased Mitochondrial Trafficking of RIP1, MLKL, Bak, Bax, and Drp1 in Hyperglycemia
2.2. Cellular Demise by Necroptosis Depends on MLKL, Bax, and Drp1 in High Glucose
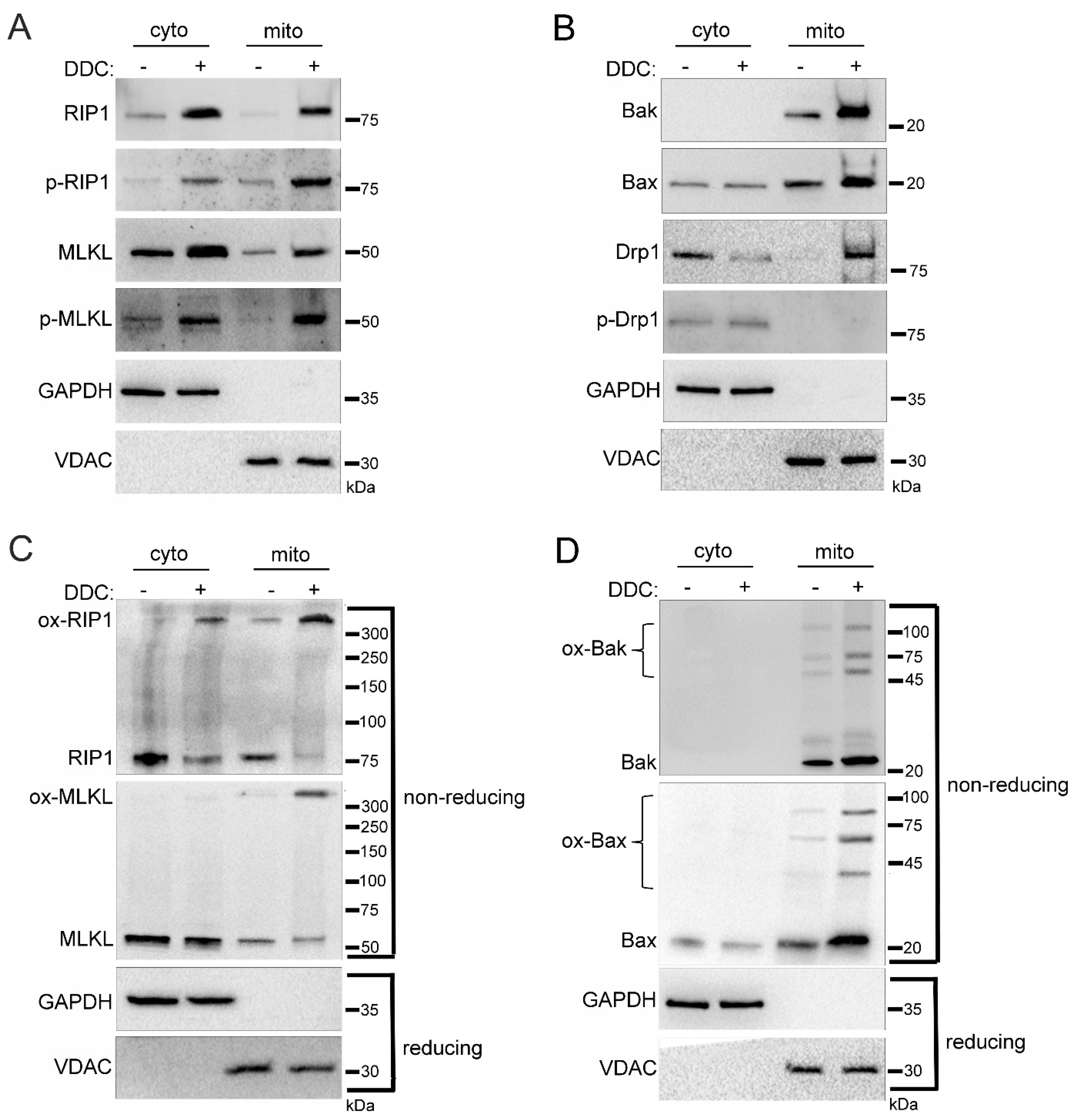
2.3. Mitochondrial Trafficking in High Glucose Depends on RIP1
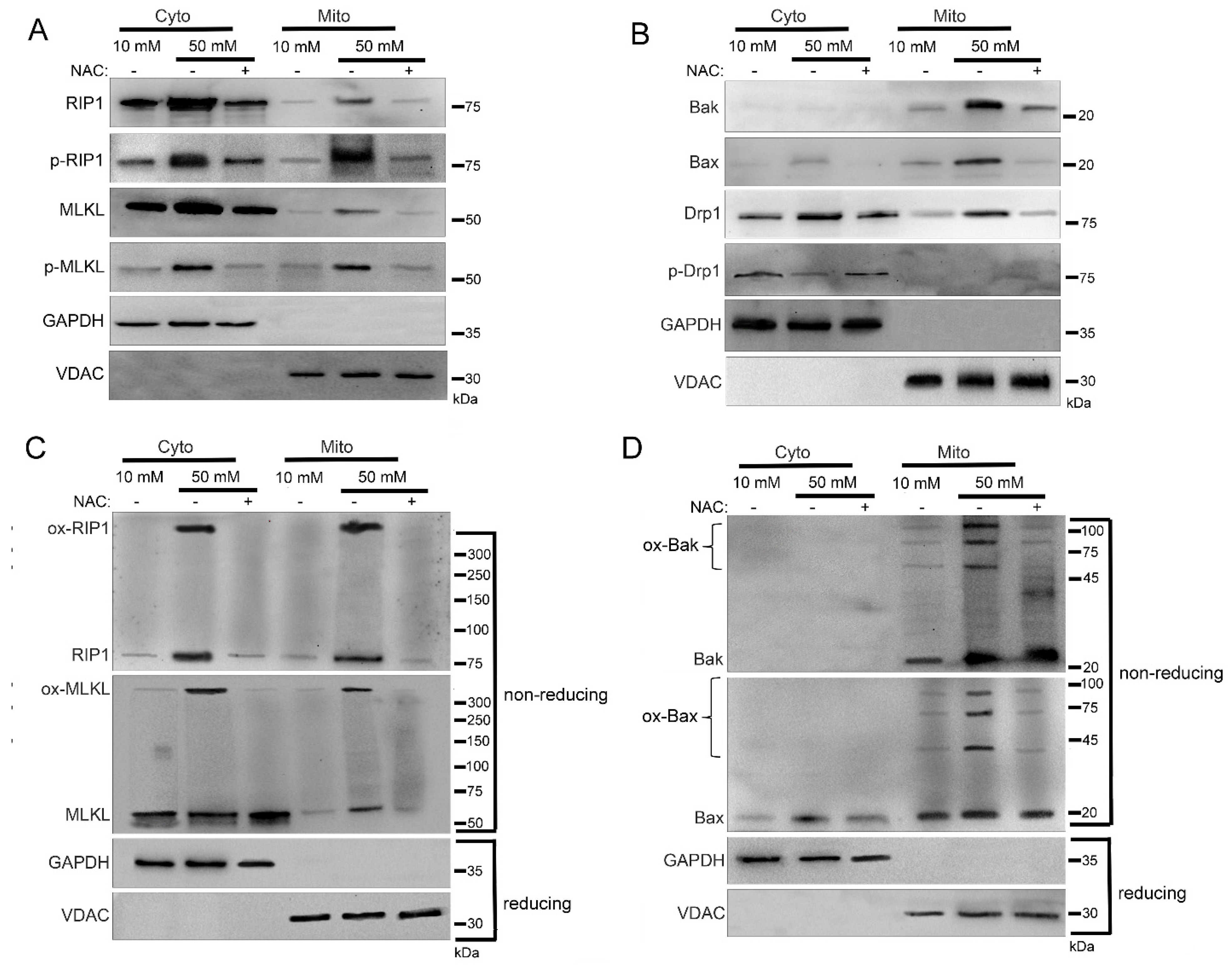
2.4. Mitochondrial Trafficking in High Glucose Depends on ROS
2.5. Sub-Mitochondrial Locations of Cell Death Factors in High Glucose
2.6. Mitochondrial Permeability and Loss of Mitochondrial Membrane Potential (Δψm) in High Glucose Depends on MLKL, Bax, and Drp1
3. Discussion
3.1. Significance of Mitochondrial Trafficking
3.2. Mitochondrial Pore Formation
3.3. Role of Drp1
4. Materials and Methods
4.1. Cell Culture and Glucose Treatments
4.2. Pharmacologic Inhibitors and Activators
4.3. Cell Fractionation
4.4. SDS-PAGE and Western Blot
4.5. WST-1 Assays
4.6. Proteinase K Assays
4.7. Fluorescence Microscopy
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in Development, Inflammation and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; vanden Berghe, T.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled Demolition at the Cellular Level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Kolbrink, B.; Riebeling, T.; Kunzendorf, U.; Krautwald, S. Plasma Membrane Pores Drive Inflammatory Cell Death. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL Forms Cation Channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef]
- Davidovich, P.; Kearney, C.; Martin, S. Inflammatory Outcomes of Apoptosis, Necrosis and Necroptosis. Biol. Chem. 2014, 395, 1163–1171. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and Its Role in Inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Zhao, H.; Jaffer, T.; Eguchi, S.; Wang, Z.; Linkermann, A.; Ma, D. Role of Necroptosis in the Pathogenesis of Solid Organ Injury. Cell Death Dis. 2015, 6, e1975. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Hackl, M.J.; Kunzendorf, U.; Walczak, H.; Krautwald, S.; Jevnikar, A.M. Necroptosis in Immunity and Ischemia-Reperfusion Injury. Am. J. Transplant. 2013, 13, 2797–2804. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Sosunov, S.A.; Shakerley, N.L.; Ten, V.S.; Ratner, A.J. Hyperglycemic Conditions Prime Cells for RIP1-Dependent Necroptosis. J. Biol. Chem. 2016, 291, 13753–13761. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Stivison, E.A.; Hod, E.A.; Spitalnik, S.L.; Cowan, P.J.; Randis, T.M.; Ratner, A.J. Human-Specific Bacterial Pore-Forming Toxins Induce Programmed Necrosis in Erythrocytes. mBio 2014, 5, e01251-14. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Stivison, E.A.; Mal-Sarkar, T.; Hooven, T.A.; Hod, E.A.; Spitalnik, S.L.; Ratner, A.J. CD59 signaling and membrane pores drive Syk-dependent erythrocyte necroptosis. Cell Death Dis. 2015, 6, e1773. [Google Scholar] [CrossRef]
- McCaig, W.D.; Hodges, A.L.; Deragon, M.A.; Haluska, R.J.; Bandyopadhyay, S.; Ratner, A.J.; Spitalnik, S.L.; Hod, E.A.; LaRocca, T.J. Storage Primes Erythrocytes for Necroptosis and Clearance. Cell. Physiol. Biochem. 2019, 53, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Deragon, M.A.; McCaig, W.D.; Patel, P.S.; Haluska, R.J.; Hodges, A.L.; Sosunov, S.A.; Murphy, M.P.; Ten, V.S.; LaRocca, T.J. Mitochondrial ROS prime the hyperglycemic shift from apoptosis to necroptosis. Cell Death Discov. 2020, 6, 132. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxidative Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Shindo, R.; Kakehashi, H.; Okumura, K.; Kumagai, Y.; Nakano, H. Critical Contribution of Oxidative Stress to TNFα-Induced Necroptosis Downstream of RIPK1 Activation. Biochem. Biophys. Res. Commun. 2013, 436, 212–216. [Google Scholar] [CrossRef]
- Qiu, X.; Zhang, Y.; Han, J. RIP3 is an upregulator of aerobic metabolism and the enhanced respiration by necrosomal RIP3 feeds back on necrosome to promote necroptosis. Cell Death Differ. 2018, 25, 821. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 Targets Pyruvate Dehydrogenase Complex to Increase Aerobic Respiration in TNF-Induced Necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, Q.; Bai, L.; Chen, W.; Wang, X.; Tellez, C.S.; Leng, S.; Padilla, M.T.; Nyunoya, T.; Belinsky, S.A.; et al. RIP1 Maintains DNA Integrity and Cell Proliferation by Regulating PGC-1α-Mediated Mitochondrial Oxidative Phosphorylation and Glycolysis. Cell Death Differ. 2014, 21, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 Autophosphorylation Is Promoted by Mitochondrial ROS and Is Essential for RIP3 Recruitment into Necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed]
- McCaig, W.D.; Patel, P.S.; Sosunov, S.A.; Shakerley, N.L.; Smiraglia, T.A.; Craft, M.M.; Walker, K.M.; Deragon, M.A.; Ten, V.S.; LaRocca, T.J. Hyperglycemia Potentiates a Shift from Apoptosis to RIP1-Dependent Necroptosis. Cell Death Discov. 2018, 4, 55. [Google Scholar] [CrossRef]
- McCaig, W.; Larocca, T. Cell Fractionation of U937 Cells by Isopycnic Density Gradient Purification. J. Vis. Exp. 2021, 2021, e62119. [Google Scholar] [CrossRef]
- van Loo, G.; Saelens, X.; Matthijssens, F.; Schotte, P.; Beyaert, R.; Declercq, W.; Vandenabeele, P. Caspases Are Not Localized in Mitochondria during Life or Death. Cell Death Differ. 2002, 9, 1207–1211. [Google Scholar] [CrossRef]
- Aslan, J.E.; Thomas, G. Death by Committee: Organellar Trafficking and Communication in Apoptosis. Traffic 2009, 10, 1390–1404. [Google Scholar] [CrossRef]
- Lackner, L.L.; Nunnari, J.M. The Molecular Mechanism and Cellular Functions of Mitochondrial Division. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 1138–1144. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Rahden-Staroń, I.; Grosicka-Macia̧g, E.; Kurpios-Piec, D.; Czeczot, H.; Grzela, T.; Szumiło, M. The Effects of Sodium Diethyldithiocarbamate in Fibroblasts V79 Cells in Relation to Cytotoxicity, Antioxidative Enzymes, Glutathione, and Apoptosis. Arch. Toxicol. 2012, 86, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.S.; Lannon, H.; Dias, L.; Zhang, J.Y.; Alper, S.L.; Pollak, M.R.; Friedman, D.J. APOL1 Kidney Risk Variants Induce Cell Death via Mitochondrial Translocation and Opening of the Mitochondrial Permeability Transition Pore. J. Am. Soc. Nephrol. 2019, 30, 2355–2368. [Google Scholar] [CrossRef] [PubMed]
- Ahting, U.; Thun, C.; Hegerl, R.; Typke, D.; Nargang, F.E.; Neupert, W.; Nussberger, S. The TOM Core Complex: The General Protein Import Pore of the Outer Membrane of Mitochondria. J. Cell Biol. 1999, 147, 959–968. [Google Scholar] [CrossRef]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate Dehydrogenase—Assembly, Regulation and Role in Human Disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef]
- Ng, F.; Tang, B.L. Pyruvate Dehydrogenase Complex (PDC) Export from the Mitochondrial Matrix. Mol. Membr. Biol. 2014, 31, 207–210. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.J.; Webb, A.I.; et al. Activation of the Pseudokinase MLKL Unleashes the Four-Helix Bundle Domain to Induce Membrane Localization and Necroptotic Cell Death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef]
- Garner, T.P.; Amgalan, D.; Reyna, D.E.; Li, S.; Kitsis, R.N.; Gavathiotis, E. Small-Molecule Allosteric Inhibitors of BAX. Nat. Chem. Biol. 2019, 15, 322–330. [Google Scholar] [CrossRef]
- Yoshida, T.; Tomioka, I.; Nagahara, T.; Holyst, T.; Sawada, M.; Hayes, P.; Gama, V.; Okuno, M.; Chen, Y.; Abe, Y.; et al. Bax-Inhibiting Peptide Derived from Mouse and Rat Ku70. Biochem. Biophys. Res. Commun. 2004, 321, 961–966. [Google Scholar] [CrossRef]
- Reyna, D.E.; Garner, T.P.; Lopez, A.; Kopp, F.; Choudhary, G.S.; Sridharan, A.; Narayanagari, S.-R.; Mitchell, K.; Dong, B.; Bartholdy, B.A.; et al. Direct Activation of BAX by BTSA1 Overcomes Apoptosis Resistance in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 490–505.e10. [Google Scholar] [CrossRef]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.M.; Baehrecke, E.H. RIP3 Finds Partners in Crime. Cell 2012, 148, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhou, Z.; Li, S.; Zhong, C.Q.; Zheng, X.; Wu, X.; Zhang, Y.; Ma, H.; Huang, D.; Li, W.; et al. Diverse Sequence Determinants Control Human and Mouse Receptor Interacting Protein 3 (RIP3) and Mixed Lineage Kinase Domain-like (MLKL) Interaction in Necroptotic Signaling. J. Biol. Chem. 2013, 288, 16247–16261. [Google Scholar] [CrossRef]
- Tanida, S.; Mizoshita, T.; Ozeki, K.; Tsukamoto, H.; Kamiya, T.; Kataoka, H.; Sakamuro, D.; Joh, T. Mechanisms of Cisplatin-Induced Apoptosis and of Cisplatin Sensitivity: Potential of BIN1 to Act as a Potent Predictor of Cisplatin Sensitivity in Gastric Cancer Treatment. Int. J. Surg. Oncol. 2012, 2012, 862879. [Google Scholar] [CrossRef]
- Karch, J.; Kanisicak, O.; Brody, M.J.; Sargent, M.A.; Michael, D.M.; Molkentin, J.D. Necroptosis Interfaces with MOMP and the MPTP in Mediating Cell Death. PLoS ONE 2015, 10, e0130520. [Google Scholar] [CrossRef]
- Ding, Y.; He, C.; Lu, S.; Wang, X.; Wang, C.; Wang, L.; Zhang, J.; Piao, M.; Chi, G.; Luo, Y.; et al. MLKL Contributes to Shikonin-Induced Glioma Cell Necroptosis via Promotion of Chromatinolysis. Cancer Lett. 2019, 467, 58–71. [Google Scholar] [CrossRef]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL Trafficking and Accumulation at the Plasma Membrane Control the Kinetics and Threshold for Necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Li, L.; Tong, A.; Zhang, Q.; Wei, Y.; Wei, X. The Molecular Mechanisms of MLKL-Dependent and MLKL-Independent Necrosis. J. Mol. Cell Biol. 2021, 13, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Molkentin, J.D. Regulated Necrotic Cell Death: The Passive Aggressive Side of Bax and Bak. Circ. Res. 2015, 116, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.-Y.; Robbins, J.; et al. Bax and Bak Function as the Outer Membrane Component of the Mitochondrial Permeability Pore in Regulating Necrotic Cell Death in Mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis Induced by RIPK3 Requires MLKL but Not Drp1. Cell Death Dis. 2014, 5, e1086-7. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and Temporal Association of Bax with Mitochondrial Fission Sites, Drp1, and Mfn2 during Apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef]
- Maes, M.E.; Grosser, J.A.; Fehrman, R.L.; Schlamp, C.L.; Nickells, R.W. Completion of BAX recruitment correlates with mitochondrial fission during apoptosis. Sci. Rep. 2019, 9, 16565. [Google Scholar] [CrossRef]
- Jenner, A.; Peña-Blanco, A.; Salvador-Gallego, R.; Ugarte-Uribe, B.; Zollo, C.; Ganief, T.; Bierlmeier, J.; Mund, M.; Lee, J.E.; Ries, J.; et al. DRP1 Interacts Directly with BAX to Induce Its Activation and Apoptosis. EMBO J. 2022, 41, e108587. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deragon, M.A.; McCaig, W.D.; Truong, P.V.; Metz, K.R.; Carron, K.A.; Hughes, K.J.; Knapp, A.R.; Dougherty, M.J.; LaRocca, T.J. Mitochondrial Trafficking of MLKL, Bak/Bax, and Drp1 Is Mediated by RIP1 and ROS which Leads to Decreased Mitochondrial Membrane Integrity during the Hyperglycemic Shift to Necroptosis. Int. J. Mol. Sci. 2023, 24, 8609. https://doi.org/10.3390/ijms24108609
Deragon MA, McCaig WD, Truong PV, Metz KR, Carron KA, Hughes KJ, Knapp AR, Dougherty MJ, LaRocca TJ. Mitochondrial Trafficking of MLKL, Bak/Bax, and Drp1 Is Mediated by RIP1 and ROS which Leads to Decreased Mitochondrial Membrane Integrity during the Hyperglycemic Shift to Necroptosis. International Journal of Molecular Sciences. 2023; 24(10):8609. https://doi.org/10.3390/ijms24108609
Chicago/Turabian StyleDeragon, Matthew A., William D. McCaig, Phillip V. Truong, Kevin R. Metz, Katherine A. Carron, Keven J. Hughes, Angeleigh R. Knapp, Molly J. Dougherty, and Timothy J. LaRocca. 2023. "Mitochondrial Trafficking of MLKL, Bak/Bax, and Drp1 Is Mediated by RIP1 and ROS which Leads to Decreased Mitochondrial Membrane Integrity during the Hyperglycemic Shift to Necroptosis" International Journal of Molecular Sciences 24, no. 10: 8609. https://doi.org/10.3390/ijms24108609
APA StyleDeragon, M. A., McCaig, W. D., Truong, P. V., Metz, K. R., Carron, K. A., Hughes, K. J., Knapp, A. R., Dougherty, M. J., & LaRocca, T. J. (2023). Mitochondrial Trafficking of MLKL, Bak/Bax, and Drp1 Is Mediated by RIP1 and ROS which Leads to Decreased Mitochondrial Membrane Integrity during the Hyperglycemic Shift to Necroptosis. International Journal of Molecular Sciences, 24(10), 8609. https://doi.org/10.3390/ijms24108609