
Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Mechanisms for Calcium Dysregulation
2.1. Presynaptic
2.2. Postsynaptic
2.3. Synaptic Plasticity
2.4. Calcium Transporters, Pumps, and Associated Membrane Channels
2.5. Astrocyte and Calcium Dynamics
2.6. Mitochondrial and Calcium Dynamics
2.7. Endoplasmic Reticulum and Calcium Dynamics
3. Novel Studies Involving Calcium Channel Modulation
3.1. Potential Therapies Targeting NMDAR
3.2. Potential Therapies Targeting VGCC
3.3. Potential Therapies Targeting TRP
3.3.1. TRP Vanilloid 4
3.3.2. TRP Cation Channel/Vanilloid Receptor
3.3.3. TRPC6
3.3.4. TRP Channel Mucolipin 1
3.4. Potential Therapies Targeting P2Y1
3.5. Potential Therapies Targeting MCU
3.6. Potential Therapies Targeting PMCA
3.7. Potential Therapies Targeting NCX3
3.8. Potential Therapies Targeting SOCE
3.9. Potential Therapies Targeting α7nAChR
3.10. Potential Therapies Targeting RyRs
3.11. Potential Therapies Targeting IP3R
3.12. Potential Therapies Targeting VDAC1
3.13. Potential Therapies Targeting CB1
3.14. Potential Therapies Targeting CaMKII and CaN
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Rosa-Neto, P.; Morais, J.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021. [Google Scholar]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, Present and Future of Therapeutic Strategies against Amyloid-β Peptides in Alzheimer’s Disease: A Systematic Review. Ageing Res. Rev. 2021, 72. [Google Scholar] [CrossRef] [PubMed]
- Prillaman, M. Alzheimer’s Drug Slows Mental Decline in Trial—But Is It a Breakthrough? Nature 2022, 610, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.B.; Shum, A.K.; Prakriya, M. Regulation of Neurogenesis by Calcium Signaling. Cell Calcium 2016, 59, 124–134. [Google Scholar] [CrossRef]
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef]
- Mateos-Aparicio, P.; Rodríguez-Moreno, A. Calcium Dynamics and Synaptic Plasticity. Adv. Exp. Med. Biol. 2020, 1131, 965–984. [Google Scholar] [CrossRef]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and Pathology of Calcium Signaling in the Brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef]
- Lee, L.; Kosuri, P.; Arancio, O. Picomolar Amyloid-β Peptides Enhance Spontaneous Astrocyte Calcium Transients. J. Alzheimer’s Dis. 2014, 38, 49–62. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium Signaling and Neurodegenerative Diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef]
- Nguyen, K.V. The Human β-Amyloid Precursor Protein: Biomolecular and Epigenetic Aspects. Biomol. Concepts 2015, 6, 11–32. [Google Scholar] [CrossRef]
- Gomez, W.; Morales, R.; Maracaja-Coutinho, V.; Parra, V.; Nassif, M. Down Syndrome and Alzheimer’s Disease: Common Molecular Traits beyond the Amyloid Precursor Protein. Aging 2020, 12, 1011–1033. [Google Scholar] [CrossRef]
- Cole, S.; Vassar, R. BACE1 Structure and Function in Health and Alzheimer’s Disease. Curr. Alzheimer Res. 2008, 5, 100–120. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Z.; Cai, F.; Zhang, M.; Wu, Y.; Zhang, J.; Song, W. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J. Neurosci. 2017, 37, 6915–6925. [Google Scholar] [CrossRef]
- Hur, J.Y. γ-Secretase in Alzheimer’s Disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-A.; Kim, H.-S.; Ha, T.-Y.; Ha, J.-W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.-P.; Park, C.-H.; Kim, S.; Baik, T.-K.; et al. Phosphorylation of Amyloid Precursor Protein (APP) at Thr668 Regulates the Nuclear Translocation of the APP Intracellular Domain and Induces Neurodegeneration. Mol. Cell Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The Amyloid-Beta Forming Tripeptide Cleavage Mechanism of γ-Secretase. Elife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s Disease-Linked Presenilin 1 Variants Elevate Abeta1-42/1-40 Ratio in Vitro and in Vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Guo, Q.; Li, H.; Gaddam, S.S.K.; Justice, N.J.; Robertson, C.S.; Zheng, H. Amyloid Precursor Protein Revisited: Neuron-Specific Expression and Highly Stable Nature of Soluble Derivatives. J. Biol. Chem. 2012, 287, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. Editorial: What Is the Physiological Function of Amyloid-Beta Protein? J. Nutr. Health Aging 2019, 23, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; Lee, B.Y.; Hane, F.T. Recent Progress in Alzheimer’s Disease Research, Part 2: Genetics and Epidemiology. J. Alzheimers Dis. 2017, 57, 317–330. [Google Scholar] [CrossRef]
- Tejada Moreno, J.A.; Villegas Lanau, A.; Madrigal Zapata, L.; Baena Pineda, A.Y.; Velez Hernandez, J.; Campo Nieto, O.; Soto Ospina, A.; Araque Marín, P.; Rishishwar, L.; Norris, E.T.; et al. Mutations in SORL1 and MTHFDL1 Possibly Contribute to the Development of Alzheimer’s Disease in a Multigenerational Colombian Family. PLoS ONE 2022, 17, e0269955. [Google Scholar] [CrossRef]
- Lesné, S.; Ming, T.K.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A Specific Amyloid-β Protein Assembly in the Brain Impairs Memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Hayley, M.; Perspicace, S.; Schulthess, T.; Seelig, J. Calcium Enhances the Proteolytic Activity of BACE1: An in Vitro Biophysical and Biochemical Characterization of the BACE1-Calcium Interaction. Biochim. Biophys. Acta 2009, 1788, 1933–1938. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 Signaling in Cortical Neurons of Knock-In Mice Expressing an Alzheimer’s-Linked Mutation in Presenilin1 Results in Exaggerated Ca2+ Signals and Altered Membrane Excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, H.S.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β Peptide Protects against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.-N.; Zhu, J.-X.; Hou, X.-H.; Shen, X.-N.; Xu, W.; Dong, Q.; Tan, L.; Yu, J.-T. Associations of Infectious Agents with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2020, 75, 299–309. [Google Scholar] [CrossRef]
- Piacentini, R.; Civitelli, L.; Ripoli, C.; Marcocci, M.E.; De Chiara, G.; Garaci, E.; Azzena, G.B.; Palamara, A.T.; Grassi, C. HSV-1 Promotes Ca2+ -Mediated APP Phosphorylation and Aβ Accumulation in Rat Cortical Neurons. Neurobiol. Aging 2011, 32, 2323.e13–2323.e26. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta Oligomers Induce Neuronal Oxidative Stress through an N-Methyl-D-Aspartate Receptor-Dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Quinn, J.F.; Harris, C.; Frahler, K.; Wadsworth, T.L.; Woltjer, R.L. L-Type Voltage-Gated Calcium Channel Blockade with Isradipine as a Therapeutic Strategy for Alzheimer’s Disease. Neurobiol. Dis. 2011, 41, 62–70. [Google Scholar] [CrossRef]
- Tong, B.C.K.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium Signaling in Alzheimer’s Disease & Therapies. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Skupin, A.; Thurley, K. Calcium Signaling: From Single Channels to Pathways. Adv. Exp. Med. Biol. 2012, 740, 531–551. [Google Scholar] [CrossRef]
- Costas-Ferreira, C.; Faro, L.R.F. Systematic Review of Calcium Channels and Intracellular Calcium Signaling: Relevance to Pesticide Neurotoxicity. Int. J. Mol. Sci. 2021, 22, 13376. [Google Scholar] [CrossRef]
- Roussarie, J.P.; Yao, V.; Rodriguez-Rodriguez, P.; Oughtred, R.; Rust, J.; Plautz, Z.; Kasturia, S.; Albornoz, C.; Wang, W.; Schmidt, E.F.; et al. Selective Neuronal Vulnerability in Alzheimer’s Disease: A Network-Based Analysis. Neuron 2020, 107, 821–835.e12. [Google Scholar] [CrossRef]
- Boopathi, S.; Garduño-Juárez, R. Calcium Inhibits Penetration of Alzheimer’s Aβ1–42 Monomers into the Membrane. Proteins Struct. Funct. Bioinform. 2022, 90, 2124–2143. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, J. Cholesterol Promotes the Interaction of Alzheimer β-Amyloid Monomer with Lipid Bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef]
- Lockhart, C.; Klimov, D.K. Calcium Enhances Binding of Aβ Monomer to DMPC Lipid Bilayer. Biophys. J. 2015, 108, 1807–1818. [Google Scholar] [CrossRef]
- Shea, T.B.; Ekinci, F.J. Biphasic Effect of Calcium Influx on Tau Phosphorylation: Phosphorylation: Biphasic Effect of Calcium Influx on Tau Phosphorylation: Involvement of Calcium-Dependent Phosphatase and Kinase Activities. J. Alzheimers Dis. 1999, 1, 353–360. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau Inhibits Mitochondrial Calcium Efflux and Makes Neurons Vulnerable to Calcium-Induced Cell Death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Leslie, S.N.; Wang, M.; Morozov, Y.M.; Yang, S.; Mentone, S.A.; Zeiss, C.; Duque, A.; Rakic, P.; Horvath, T.L.; et al. Age-Related Calcium Dysregulation Linked with Tau Pathology and Impaired Cognition in Non-Human Primates. Alzheimer’s Dement. 2021, 17, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.J.; Petersen, O.H. Neuronal Calcium Stores. Cell Calcium 1998, 24, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, J.A. Cytoplasmic Calcium Buffering: An Integrative Crosstalk. Adv. Exp. Med. Biol. 2020, 1131, 163–182. [Google Scholar] [CrossRef]
- Permyakov, E.A.; Uversky, V.N. What Is Parvalbumin For? Biomolecules 2022, 12, 656. [Google Scholar] [CrossRef]
- Mannekote Thippaiah, S.; Pradhan, B.; Voyiaziakis, E.; Shetty, R.; Iyengar, S.; Olson, C.; Tang, Y.Y. Possible Role of Parvalbumin Interneurons in Meditation and Psychiatric Illness. J. Neuropsychiatry Clin. Neurosci. 2022, 34, 113–123. [Google Scholar] [CrossRef]
- Simons, T.J.B. Calcium and Neuronal Function. Neurosurg. Rev. 1988, 11, 119–129. [Google Scholar] [CrossRef]
- Südhof, T.C. Calcium Control of Neurotransmitter Release. Cold Spring Harb. Perspect. Biol. 2012, 4, a011353. [Google Scholar] [CrossRef]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium Regulation of Neuronal Gene Expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef]
- Raymond, C.R.; Redman, S.J. Spatial Segregation of Neuronal Calcium Signals Encodes Different Forms of LTP in Rat Hippocampus. J. Physiol. 2006, 570, 97–111. [Google Scholar] [CrossRef]
- Guan, P.P.; Cao, L.L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic Dysfunction in Neurodegenerative and Neurodevelopmental Diseases: An Overview of Induced Pluripotent Stem-Cell-Based Disease Models. Open Biol. 2018, 8, 180138. [Google Scholar] [CrossRef]
- Lerdkrai, C.; Garaschuk, O. Role of Presynaptic Calcium Stores for Neural Network Dysfunction in Alzheimer’s Disease. Neural Regen. Res. 2018, 13, 977–978. [Google Scholar] [CrossRef]
- Lerdkrai, C.; Asavapanumas, N.; Brawek, B.; Kovalchuk, Y.; Mojtahedi, N.; Del Moral, M.O.; Garaschuk, O. Intracellular Ca2+ Stores Control in Vivo Neuronal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1279–E1288. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-Beta Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Westlake, T.M.; Howlett, A.C.; Bonner, T.I.; Matsuda, L.A.; Herkenham, M. Cannabinoid Receptor Binding and Messenger RNA Expression in Human Brain: An in Vitro Receptor Autoradiography and in Situ Hybridization Histochemistry Study of Normal Aged and Alzheimer’s Brains. Neuroscience 1994, 63, 637–652. [Google Scholar] [CrossRef]
- Adeoye, T.; Shah, S.I.; Demuro, A.; Rabson, D.A.; Ullah, G. Upregulated Ca2+ Release from the Endoplasmic Reticulum Leads to Impaired Presynaptic Function in Familial Alzheimer’s Disease. Cells 2022, 11, 2167. [Google Scholar] [CrossRef]
- Cooray, R.; Gupta, V.; Suphioglu, C. Current Aspects of the Endocannabinoid System and Targeted THC and CBD Phytocannabinoids as Potential Therapeutics for Parkinson’s and Alzheimer’s Diseases: A Review. Mol. Neurobiol. 2020, 57, 4878–4890. [Google Scholar] [CrossRef]
- dos Santos, R.G.; Hallak, J.E.C.; Crippa, J.A.S. Neuropharmacological Effects of the Main Phytocannabinoids: A Narrative Review. Adv. Exp. Med. Biol. 2021, 1264, 29–45. [Google Scholar] [CrossRef]
- Catani, V.M.; Gasperi, V. Assay of CB1 Receptor Binding. Methods Mol. Biol. 2016, 1412, 41–55. [Google Scholar] [CrossRef]
- Mackie, K. Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System. Handb. Exp. Pharmacol. 2005, 168, 299–325. [Google Scholar] [CrossRef]
- Freundt-Revilla, J.; Kegler, K.; Baumgärtner, W.; Tipold, A. Spatial Distribution of Cannabinoid Receptor Type 1 (CB1) in Normal Canine Central and Peripheral Nervous System. PLoS ONE 2017, 12, e0181064. [Google Scholar] [CrossRef] [PubMed]
- Monory, K.; Polack, M.; Remus, A.; Lutz, B.; Korte, M. Cannabinoid CB1 Receptor Calibrates Excitatory Synaptic Balance in the Mouse Hippocampus. J. Neurosci. 2015, 35, 3842–3850. [Google Scholar] [CrossRef] [PubMed]
- Zachariou, M.; Alexander, S.P.H.; Coombes, S.; Christodoulou, C. A Biophysical Model of Endocannabinoid-Mediated Short Term Depression in Hippocampal Inhibition. PLoS ONE 2013, 8, e58926. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Ortega-Gutiérrez, S.; Van der Stelt, M.; et al. CB1 Cannabinoid Receptors and On-Demand Defense against Excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Manuel, I.; De San Román, E.G.; Giralt, M.T.; Ferrer, I.; Rodríguez-Puertas, R. Type-1 Cannabinoid Receptor Activity during Alzheimer’s Disease Progression. J. Alzheimers Dis. 2014, 42, 761–766. [Google Scholar] [CrossRef]
- Aso, E.; Andrés-Benito, P.; Ferrer, I. Genetic Deletion of CB1 Cannabinoid Receptors Exacerbates the Alzheimer-like Symptoms in a Transgenic Animal Model. Biochem. Pharmacol. 2018, 157, 210–216. [Google Scholar] [CrossRef]
- Lee, J.H.; Agacinski, G.; Williams, J.H.; Wilcock, G.K.; Esiri, M.M.; Francis, P.T.; Wong, P.T.H.; Chen, C.P.; Lai, M.K.P. Intact Cannabinoid CB1 Receptors in the Alzheimer’s Disease Cortex. Neurochem. Int. 2010, 57, 985–989. [Google Scholar] [CrossRef]
- Ahmad, R.; Goffin, K.; Van den Stock, J.; De Winter, F.L.; Cleeren, E.; Bormans, G.; Tournoy, J.; Persoons, P.; Van Laere, K.; Vandenbulcke, M. In Vivo Type 1 Cannabinoid Receptor Availability in Alzheimer’s Disease. Eur. Neuropsychopharmacol. 2014, 24, 242–250. [Google Scholar] [CrossRef]
- Khakpai, F.; Nasehi, M.; Haeri-Rohani, A.; Eidi, A. Septo-Hippocampo-Septal Loop and Memory Formation. Basic Clin. Neurosci. 2013, 4, 5–23. [Google Scholar]
- Geula, C.; Dunlop, S.R.; Ayala, I.; Kawles, A.S.; Flanagan, M.E.; Gefen, T.; Mesulam, M.M. Basal Forebrain Cholinergic System in the Dementias: Vulnerability, Resilience, and Resistance. J. Neurochem. 2021, 158, 1394–1411. [Google Scholar] [CrossRef]
- MacHado, A.; Ferreira, D.; Grothe, M.J.; Eyjolfsdottir, H.; Almqvist, P.M.; Cavallin, L.; Lind, G.; Linderoth, B.; Seiger, Å.; Teipel, S.; et al. The Cholinergic System in Subtypes of Alzheimer’s Disease: An in Vivo Longitudinal MRI Study. Alzheimers Res. Ther. 2020, 12, 51. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Jones, S.; Sudweeks, S.; Yakel, J.L. Nicotinic Receptors in the Brain: Correlating Physiology with Function. Trends Neurosci. 1999, 22, 555–561. [Google Scholar] [CrossRef]
- Ho, T.N.T.; Abraham, N.; Lewis, R.J. Structure-Function of Neuronal Nicotinic Acetylcholine Receptor Inhibitors Derived From Natural Toxins. Front. Neurosci. 2020, 14, 609005. [Google Scholar] [CrossRef]
- Dineley, K.T. Beta-Amyloid Peptide--Nicotinic Acetylcholine Receptor Interaction: The Two Faces of Health and Disease. Front. Biosci. 2007, 12, 5030–5038. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. Beta-Amyloid(1-42) Binds to Alpha7 Nicotinic Acetylcholine Receptor with High Affinity. Implications for Alzheimer’s Disease Pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Talebi, M.; Farhoudi, M.; Golzari, S.E.J.; Sabermarouf, B.; Mahmoudi, J. Beta-Amyloid Exhibits Antagonistic Effects on Alpha 7 Nicotinic Acetylcholine Receptors in Orchestrated Manner. J. Med. Hypotheses Ideas 2014, 8, 49–52. [Google Scholar] [CrossRef]
- Lilja, A.M.; Porras, O.; Storelli, E.; Nordberg, A.; Marutle, A. Functional Interactions of Fibrillar and Oligomeric Amyloid-β with Alpha7 Nicotinic Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2011, 23, 335–347. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium Signalling and Alzheimer’s Disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of Voltage-Gated Calcium Channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Rhim, H. Effects of Amyloid-β Peptides on Voltage-Gated L-Type Ca(V)1.2 and Ca(V)1.3 Ca(2+) Channels. Mol. Cells 2011, 32, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Bobich, J.A.; Zheng, Q.; Campbell, A. Incubation of Nerve Endings with a Physiological Concentration of Abeta1-42 Activates CaV2.2(N-Type)-Voltage Operated Calcium Channels and Acutely Increases Glutamate and Noradrenaline Release. J. Alzheimers Dis. 2004, 6, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Njapo, S.A.N.; Rastogi, V.; Hedna, V.S. Taming Glutamate Excitotoxicity: Strategic Pathway Modulation for Neuroprotection. CNS Drugs 2015, 29, 153–162. [Google Scholar] [CrossRef]
- Baracaldo-Santamaría, D.; Ariza-Salamanca, D.F.; Corrales-Hernández, M.G.; Pachón-Londoño, M.J.; Hernandez-Duarte, I.; Calderon-Ospina, C.A. Revisiting Excitotoxicity in Traumatic Brain Injury: From Bench to Bedside. Pharmaceutics 2022, 14, 152. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in Acute Stroke: Targeting Excitotoxicity, Oxidative and Nitrosative Stress, and Inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Ariza-Salamanca, D.F.; Corrales-Hernández, M.G.; Pachón-Londoño, M.J.; Hernández-Duarte, I. Molecular and Cellular Mechanisms Leading to Catatonia: An Integrative Approach from Clinical and Preclinical Evidence. Front. Mol. Neurosci. 2022, 15, 993671. [Google Scholar] [CrossRef]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of Action of Memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Lladó, A.; Rami, L. Memantine: Targeting Glutamate Excitotoxicity in Alzheimer’s Disease and Other Dementias. Am. J. Alzheimers Dis. Other Demen. 2005, 20, 77–85. [Google Scholar] [CrossRef]
- Goussakov, I.; Miller, M.B.; Stutzmann, G.E. NMDA-Mediated Ca(2+) Influx Drives Aberrant Ryanodine Receptor Activation in Dendrites of Young Alzheimer’s Disease Mice. J. Neurosci. 2010, 30, 12128–12137. [Google Scholar] [CrossRef]
- Nimmrich, V.; Grimm, C.; Draguhn, A.; Barghorn, S.; Lehmann, A.; Schoemaker, H.; Hillen, H.; Gross, G.; Ebert, U.; Bruehl, C. Amyloid β Oligomers (Aβ1–42 Globulomer) Suppress Spontaneous Synaptic Activity by Inhibition of P/Q-Type Calcium Currents. J. Neurosci. 2008, 28, 788. [Google Scholar] [CrossRef]
- Juhász, G.; Barkóczi, B.; Vass, G.; Datki, Z.; Hunya, Á.; Fülöp, L.; Budai, D.; Penke, B.; Szegedi, V. Fibrillar Abeta (1-42) Enhances NMDA Receptor Sensitivity via the Integrin Signaling Pathway. J. Alzheimers Dis. 2010, 19, 1055–1067. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Lalanne, T.; Oyrer, J.; Farrant, M.; Sjöström, P.J. Synapse Type-Dependent Expression of Calcium-Permeable AMPA Receptors. Front. Synaptic Neurosci. 2018, 10, 34. [Google Scholar] [CrossRef]
- Rozov, A.; Sprengel, R.; Seeburg, P.H. GluA2-Lacking AMPA Receptors in Hippocampal CA1 Cell Synapses: Evidence from Gene-Targeted Mice. Front. Mol. Neurosci. 2012, 5, 22. [Google Scholar] [CrossRef]
- Stubblefield, E.A.; Benke, T.A. Distinct AMPA-Type Glutamatergic Synapses in Developing Rat CA1 Hippocampus. J. Neurophysiol. 2010, 104, 1899–1912. [Google Scholar] [CrossRef]
- Park, P.; Kang, H.; Georgiou, J.; Zhuo, M.; Kaang, B.K.; Collingridge, G.L. Further Evidence That CP-AMPARs Are Critically Involved in Synaptic Tag and Capture at Hippocampal CA1 Synapses. Mol. Brain 2021, 14, 26. [Google Scholar] [CrossRef]
- Whitcomb, D.J.; Hogg, E.L.; Regan, P.; Piers, T.; Narayan, P.; Whitehead, G.; Winters, B.L.; Kim, D.H.; Kim, E.; St George-Hyslop, P.; et al. Intracellular Oligomeric Amyloid-Beta Rapidly Regulates GluA1 Subunit of AMPA Receptor in the Hippocampus. Sci. Rep. 2015, 5, 10934. [Google Scholar] [CrossRef] [PubMed]
- Findley, C.A.; Bartke, A.; Hascup, K.N.; Hascup, E.R. Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics During Alzheimer’s Disease Progression. ASN Neuro 2019, 11, 1759091419855541. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimers Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [PubMed]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Lu, R.; He, Q.; Wang, J. TRPC Channels and Alzheimer’s Disease. Adv. Exp. Med. Biol. 2017, 976, 73–83. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X.; Gao, Q.; Xu, H. TRPML1: An Ion Channel in the Lysosome. Handb. Exp. Pharmacol. 2014, 222, 631–645. [Google Scholar] [CrossRef]
- Putney, J.W. Type 3 Inositol 1,4,5-Trisphosphate Receptor and Capacitative Calcium Entry. Cell Calcium 1997, 21, 257–261. [Google Scholar] [CrossRef]
- Shen, H.; Pan, J.; Pan, L.; Zhang, N. TRPC6 Inhibited NMDA Current in Cultured Hippocampal Neurons. Neuromol. Med. 2013, 15, 389–395. [Google Scholar] [CrossRef]
- Tai, Y.; Feng, S.; Ge, R.; Du, W.; Zhang, X.; He, Z.; Wang, Y. TRPC6 Channels Promote Dendritic Growth via the CaMKIV-CREB Pathway. J. Cell Sci. 2008, 121, 2301–2307. [Google Scholar] [CrossRef]
- Prikhodko, V.; Chernyuk, D.; Sysoev, Y.; Zernov, N.; Okovityi, S.; Popugaeva, E. Potential Drug Candidates to Treat TRPC6 Channel Deficiencies in the Pathophysiology of Alzheimer’s Disease and Brain Ischemia. Cells 2020, 9, 2351. [Google Scholar] [CrossRef]
- Lessard, C.B.; Lussier, M.P.; Cayouette, S.; Bourque, G.; Boulay, G. The Overexpression of Presenilin2 and Alzheimer’s-Disease-Linked Presenilin2 Variants Influences TRPC6-Enhanced Ca2+ Entry into HEK293 Cells. Cell Signal 2005, 17, 437–445. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of Intracellular Calcium Signaling in Alzheimer’s Disease. Antioxid. Redox Signal. 2018, 29, 1176–1188. [Google Scholar] [CrossRef]
- Ho, R.; Ortiz, D. Amyloid-Beta Promotes Calcium Influx and Neurodegeneration via Stimulation of L Voltage-Sensitive Calcium Channels Rather than NMDA Channels in Cultured Neurons. J. Alzheimers Dis. 2001, 3, 479–483. [Google Scholar] [CrossRef]
- Ekinci, F.J.; Malik, K.U.; Shea, T.B. Activation of the L Voltage-Sensitive Calcium Channel by Mitogen-Activated Protein (MAP) Kinase Following Exposure of Neuronal Cells to Beta-Amyloid. MAP Kinase Mediates Beta-Amyloid-Induced Neurodegeneration. J. Biol. Chem. 1999, 274, 30322–30327. [Google Scholar] [CrossRef]
- Coon, A.L.; Wallace, D.R.; MacTutus, C.F.; Booze, R.M. L-Type Calcium Channels in the Hippocampus and Cerebellum of Alzheimer’s Disease Brain Tissue. Neurobiol. Aging 1999, 20, 597–603. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative Stress in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Davis, D.R.; Anderton, B.H.; Brion, J.P.; Reynolds, C.H.; Hanger, D.P. Oxidative Stress Induces Dephosphorylation of Tau in Rat Brain Primary Neuronal Cultures. J. Neurochem. 1997, 68, 1590–1597. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef]
- Uryash, A.; Flores, V.; Adams, J.A.; Allen, P.D.; Lopez, J.R. Memory and Learning Deficits Are Associated With Ca2+ Dyshomeostasis in Normal Aging. Front. Aging Neurosci. 2020, 12, 224. [Google Scholar] [CrossRef]
- Chapman, P.F.; White, G.L.; Jones, M.W.; Cooper-Blacketer, D.; Marshall, V.J.; Irizarry, M.; Younkin, L.; Good, M.A.; Bliss, T.V.P.; Hyman, B.T.; et al. Impaired Synaptic Plasticity and Learning in Aged Amyloid Precursor Protein Transgenic Mice. Nat. Neurosci. 1999, 2, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Zhang, J.; Chen, S.; Huang, Y.; Chen, W.; He, L.; Zhang, Y. Role of Calcium Homeostasis in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2022, 18, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Cárdenas, C.; Mei, L.; Cheung, K.H.; Foskett, J.K. Constitutive CAMP Response Element Binding Protein (CREB) Activation by Alzheimer’s Disease Presenilin-Driven Inositol Trisphosphate Receptor (InsP3R) Ca2+ Signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 13293–13298. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Goncalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.P.; et al. Impaired Learning and LTP in Mice Expressing the Carboxy Terminus of the Alzheimer Amyloid Precursor Protein. Nature 1997, 387, 500–505. [Google Scholar] [CrossRef]
- Chakroborty, S.; Kim, J.; Schneider, C.; Jacobson, C.; Molgó, J.; Stutzmann, G.E. Early Presynaptic and Postsynaptic Calcium Signaling Abnormalities Mask Underlying Synaptic Depression in Presymptomatic Alzheimer’s Disease Mice. J. Neurosci. 2012, 32, 8341–8353. [Google Scholar] [CrossRef]
- Ghosh, A.; Giese, K.P. Calcium/Calmodulin-Dependent Kinase II and Alzheimer’s Disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef]
- McKee, A.C.; Kosik, K.S.; Kennedy, M.B.; Kowall, N.W. Hippocampal Neurons Predisposed to Neurofibrillary Tangle Formation Are Enriched in Type II Calcium/Calmodulin-Dependent Protein Kinase. J. Neuropathol. Exp. Neurol. 1990, 49, 49–63. [Google Scholar] [CrossRef]
- Mah, V.H.; Eskin, T.A.; Kazee, A.M.; Lapham, L.; Higgins, G.A. In Situ Hybridization of Calcium/Calmodulin Dependent Protein Kinase II and Tau MRNAs; Species Differences and Relative Preservation in Alzheimer’s Disease. Brain Res. Mol. Brain Res. 1992, 12, 85–94. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yamauchi, E.; Taniguchi, H.; Ono, T.; Miyamoto, E. Phosphorylation of Microtubule-Associated Protein Tau by Ca2+/Calmodulin-Dependent Protein Kinase II in Its Tubulin Binding Sites. Arch. Biochem. Biophys. 2002, 408, 255–262. [Google Scholar] [CrossRef]
- Reese, L.C.; Zhang, W.R.; Dineley, K.T.; Kayed, R.; Taglialatela, G. Selective Induction of Calcineurin Activity and Signaling by Oligomeric Amyloid Beta. Aging Cell 2008, 7, 824–835. [Google Scholar] [CrossRef]
- Dineley, K.T.; Hogan, D.; Zhang, W.R.; Taglialatela, G. Acute Inhibition of Calcineurin Restores Associative Learning and Memory in Tg2576 APP Transgenic Mice. Neurobiol. Learn. Mem. 2007, 88, 217–224. [Google Scholar] [CrossRef]
- Cullen, W.K.; Suh, Y.H.; Anwyl, R.; Rowan, M.J. Block of LTP in Rat Hippocampus in Vivo by Beta-Amyloid Precursor Protein Fragments. Neuroreport 1997, 8, 3213–3217. [Google Scholar] [CrossRef]
- Freir, D.B.; Holscher, C.; Herron, C.E. Blockade of Long-Term Potentiation by Beta-Amyloid Peptides in the CA1 Region of the Rat Hippocampus in Vivo. J. Neurophysiol. 2001, 85, 708–713. [Google Scholar] [CrossRef]
- Chen, Q.S.; Wei, W.Z.; Shimahara, T.; Xie, C.W. Alzheimer Amyloid β-Peptide Inhibits the Late Phase of Long-Term Potentiation through Calcineurin-Dependent Mechanisms in the Hippocampal Dentate Gyrus. Neurobiol. Learn. Mem. 2002, 77, 354–371. [Google Scholar] [CrossRef]
- Zhang, J.F.; Qi, J.S.; Qiao, J.T. Protein Kinase C Mediates Amyloid Beta-Protein Fragment 31-35-Induced Suppression of Hippocampal Late-Phase Long-Term Potentiation in Vivo. Neurobiol. Learn. Mem. 2009, 91, 226–234. [Google Scholar] [CrossRef]
- Yeung, J.H.Y.; Walby, J.L.; Palpagama, T.H.; Turner, C.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Glutamatergic Receptor Expression Changes in the Alzheimer’s Disease Hippocampus and Entorhinal Cortex. Brain Pathol. 2021, 31, e13005. [Google Scholar] [CrossRef]
- Zhao, D.; Watson, J.B.; Xie, C.W. Amyloid Beta Prevents Activation of Calcium/Calmodulin-Dependent Protein Kinase II and AMPA Receptor Phosphorylation during Hippocampal Long-Term Potentiation. J. Neurophysiol. 2004, 92, 2853–2858. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Lüscher, C.; Huber, K.M. Group 1 MGluR-Dependent Synaptic Long-Term Depression: Mechanisms and Implications for Circuitry and Disease. Neuron 2010, 65, 445–459. [Google Scholar] [CrossRef]
- Hovelso, N.; Sotty, F.P.; Montezinho, L.; S. Pinheiro, P.; F. Herrik, K.; Mork, A. Therapeutic Potential of Metabotropic Glutamate Receptor Modulators. Curr. Neuropharmacol. 2012, 10, 12–48. [Google Scholar] [CrossRef]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease Through Prion Protein and MGluR5. Adv. Pharmacol. 2018, 82, 293–323. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elrahman, K.S.; Ferguson, S.S.G. Noncanonical Metabotropic Glutamate Receptor 5 Signaling in Alzheimer’s Disease. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Fraser, P.E.; Westaway, D.; St George-Hyslop, P.H.; Ehrlich, M.E.; Gandy, S. Group II Metabotropic Glutamate Receptor Stimulation Triggers Production and Release of Alzheimer’s Amyloid(Beta)42 from Isolated Intact Nerve Terminals. J. Neurosci. 2010, 30, 3870–3875. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.E.; Wang, M.; Galvin, V.C.; Lightbourne, T.C.; Conn, P.J.; Arnsten, A.F.T.; Paspalas, C.D. MGluR2 versus MGluR3 Metabotropic Glutamate Receptors in Primate Dorsolateral Prefrontal Cortex: Postsynaptic MGluR3 Strengthen Working Memory Networks. Cereb. Cortex. 2018, 28, 974–987. [Google Scholar] [CrossRef]
- Gu, Z.; Cheng, J.; Zhong, P.; Qin, L.; Liu, W.; Yan, Z. Aβ Selectively Impairs MGluR7 Modulation of NMDA Signaling in Basal Forebrain Cholinergic Neurons: Implication in Alzheimer’s Disease. J. Neurosci. 2014, 34, 13614–13628. [Google Scholar] [CrossRef]
- Woo, M.S.; Ufer, F.; Rothammer, N.; Di Liberto, G.; Binkle, L.; Haferkamp, U.; Sonner, J.K.; Engler, J.B.; Hornig, S.; Bauer, S.; et al. Neuronal Metabotropic Glutamate Receptor 8 Protects against Neurodegeneration in CNS Inflammation. J. Exp. Med. 2021, 218, e20201290. [Google Scholar] [CrossRef]
- Iscru, E.; Goddyn, H.; Ahmed, T.; Callaerts-Vegh, Z.; D’Hooge, R.; Balschun, D. Improved Spatial Learning Is Associated with Increased Hippocampal but Not Prefrontal Long-Term Potentiation in MGluR4 Knockout Mice. Genes Brain Behav. 2013, 12, 615–625. [Google Scholar] [CrossRef]
- Guerini, D. The Ca2+ Pumps and the Na+/Ca2+ Exchangers. Biometals 1998, 11, 319–330. [Google Scholar] [CrossRef]
- Wu, X.; Weng, L.; Zhang, J.; Liu, X.; Huang, J. The Plasma Membrane Calcium ATPases in Calcium Signaling Network. Curr. Protein Pept. Sci. 2018, 19, 813–822. [Google Scholar] [CrossRef]
- Kip, S.N.; Strehler, E.E. Rapid Downregulation of NCX and PMCA in Hippocampal Neurons Following H2O2 Oxidative Stress. Ann. N. Y. Acad. Sci. 2007, 1099, 436–439. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium Dysregulation and Homeostasis of Neural Calcium in the Molecular Mechanisms of Neurodegenerative Diseases Provide Multiple Targets for Neuroprotection. Antioxid. Redox. Signal 2011, 14, 1275. [Google Scholar] [CrossRef]
- Guan, P.P.; Cao, L.L.; Yang, Y.; Wang, P. Calcium Ions Aggravate Alzheimer’s Disease Through the Aberrant Activation of Neuronal Networks, Leading to Synaptic and Cognitive Deficits. Front. Mol. Neurosci. 2021, 14, 298. [Google Scholar] [CrossRef]
- Pannaccione, A.; Piccialli, I.; Secondo, A.; Ciccone, R.; Molinaro, P.; Boscia, F.; Annunziato, L. The Na+/Ca2+exchanger in Alzheimer’s Disease. Cell Calcium 2020, 87, 102190. [Google Scholar] [CrossRef]
- Lytton, J. Na+/Ca2+ Exchangers: Three Mammalian Gene Families Control Ca2+ Transport. Biochem. J. 2007, 406, 365–382. [Google Scholar] [CrossRef]
- Piccialli, I.; Ciccone, R.; Secondo, A.; Boscia, F.; Tedeschi, V.; de Rosa, V.; Cepparulo, P.; Annunziato, L.; Pannaccione, A. The Na+/Ca2+ Exchanger 3 Is Functionally Coupled With the NaV1.6 Voltage-Gated Channel and Promotes an Endoplasmic Reticulum Ca2+ Refilling in a Transgenic Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 775271. [Google Scholar] [CrossRef]
- Moriguchi, S.; Kita, S.; Fukaya, M.; Osanai, M.; Inagaki, R.; Sasaki, Y.; Izumi, H.; Horie, K.; Takeda, J.; Saito, T.; et al. Reduced Expression of Na+/Ca2+ Exchangers Is Associated with Cognitive Deficits Seen in Alzheimer’s Disease Model Mice. Neuropharmacology 2018, 131, 291–303. [Google Scholar] [CrossRef]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef]
- Sugunan, S.; Nampoothiri, S.; Garg, T.; Krishnamurthy, R. Role of KCa3.1 Channels in CNS Diseases: A Concise Review. CNS Neurol. Disord. Drug Targets 2016, 15, 1299–1305. [Google Scholar] [CrossRef]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-Activated Potassium Channels: Implications for Aging and Age-Related Neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef]
- Sancho, M.; Kyle, B.D. The Large-Conductance, Calcium-Activated Potassium Channel: A Big Key Regulator of Cell Physiology. Front. Physiol. 2021, 12, 750615. [Google Scholar] [CrossRef]
- Tazerart, S.; Blanchard, M.G.; Miranda-Rottmann, S.; Mitchell, D.E.; Navea Pina, B.; Thomas, C.I.; Kamasawa, N.; Araya, R. Selective Activation of BK Channels in Small-Headed Dendritic Spines Suppresses Excitatory Postsynaptic Potentials. J. Physiol. 2022, 600, 2165–2187. [Google Scholar] [CrossRef] [PubMed]
- Bock, T.; Stuart, G.J. The Impact of BK Channels on Cellular Excitability Depends on Their Subcellular Location. Front. Cell Neurosci. 2016, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Pedarzani, P.; Stocker, M. Molecular and Cellular Basis of Small--and Intermediate-Conductance, Calcium-Activated Potassium Channel Function in the Brain. Cell Mol. Life Sci. 2008, 65, 3196–3217. [Google Scholar] [CrossRef] [PubMed]
- Blank, T.; Nijholt, I.; Kye, M.J.; Radulovic, J.; Spiess, J. Small-Conductance, Ca2+-Activated K+ Channel SK3 Generates Age-Related Memory and LTP Deficits. Nat. Neurosci. 2003, 6, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Benninger, F.; Yaari, Y. Role of Small Conductance Ca2+-Activated K+ Channels in Controlling CA1 Pyramidal Cell Excitability. J. Neurosci. 2014, 34, 8219–8230. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamamoto, R.; Kato, N. Amyloid β and Amyloid Precursor Protein Synergistically Suppress Large-Conductance Calcium-Activated Potassium Channel in Cortical Neurons. Front. Aging Neurosci. 2021, 13, 660319. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ueta, Y.; Wang, L.; Yamamoto, R.; Inoue, N.; Inokuchi, K.; Aiba, A.; Yonekura, H.; Kato, N. Suppression of a Neocortical Potassium Channel Activity by Intracellular Amyloid-β and Its Rescue with Homer1a. J. Neurosci. 2011, 31, 11100–11109. [Google Scholar] [CrossRef]
- Hakim, M.A.; Behringer, E.J.; Aguayo, L. Development of Alzheimer’s Disease Progressively Alters Sex-Dependent KCa and Sex-Independent KIR Channel Function in Cerebrovascular Endothelium. J. Alzheimers Dis. 2020, 76, 1423–1442. [Google Scholar] [CrossRef]
- John, C.M.; Khaddaj Mallat, R.; Mishra, R.C.; George, G.; Singh, V.; Turnbull, J.D.; Umeshappa, C.S.; Kendrick, D.J.; Kim, T.; Fauzi, F.M.; et al. SKA-31, an Activator of Ca2+-Activated K+ Channels, Improves Cardiovascular Function in Aging. Pharmacol. Res. 2020, 151, 104539. [Google Scholar] [CrossRef]
- Yang, X.; Wang, G.; Cao, T.; Zhang, L.; Ma, Y.; Jiang, S.; Teng, X.; Sun, X. Large-Conductance Calcium-Activated Potassium Channels Mediate Lipopolysaccharide-Induced Activation of Murine Microglia. J. Biol. Chem. 2019, 294, 12921–12932. [Google Scholar] [CrossRef]
- Maezawa, I.; Jenkins, D.P.; Jin, B.E.; Wulff, H. Microglial KCa3.1 Channels as a Potential Therapeutic Target for Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012, 2012, 868972. [Google Scholar] [CrossRef]
- Hwang, S.M.; Lee, J.Y.; Park, C.K.; Kim, Y.H. The Role of TRP Channels and PMCA in Brain Disorders: Intracellular Calcium and PH Homeostasis. Front. Cell Dev. Biol. 2021, 9, 584388. [Google Scholar] [CrossRef]
- Boczek, T.; Radzik, T.; Ferenc, B.; Zylinska, L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. Int. J. Mol. Sci. 2019, 20, 6338. [Google Scholar] [CrossRef]
- Lin, X.; Brunk, M.G.K.; Yuanxiang, P.; Curran, A.W.; Zhang, E.; Stöber, F.; Goldschmidt, J.; Gundelfinger, E.D.; Vollmer, M.; Happel, M.F.K.; et al. Neuroplastin Expression Is Essential for Hearing and Hair Cell PMCA Expression. Brain Struct. Funct. 2021, 226, 1533–1551. [Google Scholar] [CrossRef]
- Mata, A.M.; Sepulveda, M.R. Plasma Membrane Ca2+-ATPases in the Nervous System during Development and Ageing. World J. Biol. Chem. 2010, 1, 229. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef]
- Berrocal, M.; Marcos, D.; Sepúlveda, M.R.; Pérez, M.; Ávila, J.; Mata, A.M. Altered Ca2+ Dependence of Synaptosomal Plasma Membrane Ca2+-ATPase in Human Brain Affected by Alzheimer’s Disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef]
- Sepúlveda, M.R.; Berrocal-Carrillo, M.; Gasset, M.; Mata, A.M. The Plasma Membrane Ca2+-ATPase Isoform 4 Is Localized in Lipid Rafts of Cerebellum Synaptic Plasma Membranes. J. Biol. Chem. 2006, 281, 447–453. [Google Scholar] [CrossRef]
- Semyanov, A.; Henneberger, C.; Agarwal, A. Making Sense of Astrocytic Calcium Signals—From Acquisition to Interpretation. Nat. Rev. Neurosci. 2020, 21, 551–564. [Google Scholar] [CrossRef]
- Guerra-Gomes, S.; Sousa, N.; Pinto, L.; Oliveira, J.F. Functional Roles of Astrocyte Calcium Elevations: From Synapses to Behavior. Front. Cell Neurosci. 2018, 11, 427. [Google Scholar] [CrossRef]
- Lia, A.; Henriques, V.J.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell Neurosci. 2021, 15, 673433. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Perea, G.; Maglio, L.; Pastor, J.; García De Sola, R.; Araque, A. Astrocyte Calcium Signal and Gliotransmission in Human Brain Tissue. Cereb. Cortex. 2013, 23, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Reyes, R.C.; Parpura, V. TRP Channels Coordinate Ion Signalling in Astroglia. Rev. Physiol. Biochem. Pharmacol. 2014, 166. [Google Scholar] [CrossRef]
- Jackson, J.G.; Robinson, M.B. Regulation of Mitochondrial Dynamics in Astrocytes: Mechanisms, Consequences, and Unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y. Astrocytic Ca2+ Signaling Mediated by the Endoplasmic Reticulum in Health and Disease. J. Pharmacol. Sci. 2020, 144, 83–88. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Bégard, S.; Pythoud, C.; Rey, M.; et al. Tau Accumulation in Astrocytes of the Dentate Gyrus Induces Neuronal Dysfunction and Memory Deficits in Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β Impairs the Phagocytosis of Dystrophic Synapses by Astrocytes in Alzheimer’s Disease. Glia 2021, 69, 997–1011. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor Signalling Mediates Astrocytic Hyperactivity in Vivo in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 Receptor Blockade Normalizes Network Dysfunction and Cognition in an Alzheimer’s Disease Model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef]
- Sompol, P.; Norris, C.M. Ca2+, Astrocyte Activation and Calcineurin/NFAT Signaling in Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 199. [Google Scholar] [CrossRef]
- Abdul, H.M.; Sama, M.A.; Furman, J.L.; Mathis, D.M.; Beckett, T.L.; Weidner, A.M.; Patel, E.S.; Baig, I.; Murphy, M.P.; LeVine, H.; et al. Cognitive Decline in Alzheimer’s Disease Is Associated with Selective Changes in Calcineurin/NFAT Signaling. J. Neurosci. 2009, 29, 12957–12969. [Google Scholar] [CrossRef]
- Brezovakova, V.; Sykova, E.; Jadhav, S. Astrocytes Derived from Familial and Sporadic Alzheimer’s Disease IPSCs Show Altered Calcium Signaling and Respond Differently to Misfolded Protein Tau. Cells 2022, 11, 1429. [Google Scholar] [CrossRef]
- Mustaly-Kalimi, S.; Littlefield, A.M.; Stutzmann, G.E. Calcium Signaling Deficits in Glia and Autophagic Pathways Contributing to Neurodegenerative Disease. Antioxid. Redox. Signal 2018, 29, 1158–1175. [Google Scholar] [CrossRef]
- Pra, I.D.; Chiarini, A.; Nemeth, E.F.; Armato, U.; Whitfield, J.F. Roles of Ca2+ and the Ca2+-Sensing Receptor (CASR) in the Expression of Inducible NOS (Nitric Oxide Synthase)-2 and Its BH4 (Tetrahydrobiopterin)-Dependent Activation in Cytokine-Stimulated Adult Human Astrocytes. J. Cell Biochem. 2005, 96, 428–438. [Google Scholar] [CrossRef]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The Calcium-Sensing Receptor in Physiology and in Calcitropic and Noncalcitropic Diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef]
- Gorvin, C.M. Calcium-Sensing Receptor Signaling—How Human Disease Informs Biology. Curr. Opin. Endocr. Metab. Res. 2021, 16, 10–18. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9036. [Google Scholar] [CrossRef]
- Pra, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.; Armato, U. Calcium-Sensing Receptors of Human Astrocyte-Neuron Teams: Amyloid-β-Driven Mediators and Therapeutic Targets of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 353–364. [Google Scholar] [CrossRef]
- Feng, C.; Bao, X.; Shan, L.; Ling, Y.; Ding, Y.; Wang, J.; Cao, Y.; Wang, Q.; Cui, W.; Xu, S. Calcium-Sensing Receptor Mediates β-Amyloid-Induced Synaptic Formation Impairment and Cognitive Deficits via Regulation of Cytosolic Phospholipase A2/Prostaglandin E2 Metabolic Pathway. Front. Aging Neurosci. 2020, 12, 144. [Google Scholar] [CrossRef]
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased Calcium-Sensing Receptor Immunoreactivity in the Hippocampus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-Sensing Receptor Antagonist (Calcilytic) NPS 2143 Specifically Blocks the Increased Secretion of Endogenous Aβ42 Prompted by Exogenous Fibrillary or Soluble Aβ25-35 in Human Cortical Astrocytes and Neurons-Therapeutic Relevance to Alzheimer’s Dise. Biochim. Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Liu, D.; Pra, I.D. Calcium-Sensing Receptor Antagonist NPS 2143 Restores Amyloid Precursor Protein Physiological Non-Amyloidogenic Processing in Aβ-Exposed Adult Human Astrocytes. Sci. Rep. 2017, 7, 1277. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells 2020, 9, 1386. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The Role of Transient Receptor Potential Cation Channels in Ca2+ Signaling. Cold Spring Harb Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Gudermann, T. TRPC6. Handb. Exp. Pharmacol. 2007, 179, 125–141. [Google Scholar] [CrossRef]
- Zhou, J.; Du, W.; Zhou, K.; Tai, Y.; Yao, H.; Jia, Y.; Ding, Y.; Wang, Y. Critical Role of TRPC6 Channels in the Formation of Excitatory Synapses. Nat. Neurosci. 2008, 11, 741–743. [Google Scholar] [CrossRef]
- Jia, Y.; Zhou, J.; Tai, Y.; Wang, Y. TRPC Channels Promote Cerebellar Granule Neuron Survival. Nat. Neurosci. 2007, 10, 559–567. [Google Scholar] [CrossRef]
- Lu, R.; Wang, J.; Tao, R.; Wang, J.; Zhu, T.; Guo, W.; Sun, Y.; Li, H.; Gao, Y.; Zhang, W.; et al. Reduced TRPC6 MRNA Levels in the Blood Cells of Patients with Alzheimer’s Disease and Mild Cognitive Impairment. Mol. Psychiatry 2018, 23, 767–776. [Google Scholar] [CrossRef]
- Chen, J.M.; Li, Q.W.; Liu, J.S.; Jiang, G.X.; Liu, J.R.; Chen, S.D.; Cheng, Q. TRPC6 MRNA Levels in Peripheral Leucocytes of Patients with Alzheimer’s Disease and Mild Cognitive Impairment: A Case-Control Study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 279–284. [Google Scholar] [CrossRef]
- Beskina, O.; Miller, A.; Mazzocco-Spezzia, A.; Pulina, M.V.; Golovina, V.A. Mechanisms of Interleukin-1beta-Induced Ca2+ Signals in Mouse Cortical Astrocytes: Roles of Store- and Receptor-Operated Ca2+ Entry. Am. J. Physiol. Cell Physiol. 2007, 293, C1103–C1111. [Google Scholar] [CrossRef]
- Tu, P.; Gibon, J.; Bouron, A. The TRPC6 Channel Activator Hyperforin Induces the Release of Zinc and Calcium from Mitochondria. J. Neurochem. 2010, 112, 204–213. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, Y.; Shu, H.; Xie, B.; Tao, Y.; Yuan, Y.; Shang, Y.; Yuan, S.; Zhang, J. Hyperforin Promotes Post-Stroke Neuroangiogenesis via Astrocytic IL-6-Mediated Negative Immune Regulation in the Ischemic Brain. Front. Cell Neurosci. 2019, 13, 201. [Google Scholar] [CrossRef]
- Bernal-Chico, A.; Tepavcevic, V.; Manterola, A.; Utrilla, C.; Matute, C.; Mato, S. Endocannabinoid Signaling in Brain Diseases: Emerging Relevance of Glial Cells. Glia 2023, 71, 103–126. [Google Scholar] [CrossRef]
- Gutiérrez-Rodríguez, A.; Bonilla-Del Río, I.; Puente, N.; Gómez-Urquijo, S.M.; Fontaine, C.J.; Egaña-Huguet, J.; Elezgarai, I.; Ruehle, S.; Lutz, B.; Robin, L.M.; et al. Localization of the Cannabinoid Type-1 Receptor in Subcellular Astrocyte Compartments of Mutant Mouse Hippocampus. Glia 2018, 66, 1417–1431. [Google Scholar] [CrossRef]
- Achicallende, S.; Bonilla-Del Río, I.; Serrano, M.; Mimenza, A.; Lekunberri, L.; Anaut-Lusar, I.; Puente, N.; Gerrikagoitia, I.; Grandes, P. GLAST versus GFAP as Astroglial Marker for the Subcellular Study of Cannabinoid CB1 Receptors in Astrocytes. Histochem. Cell Biol. 2022, 158, 561–569. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. Endocannabinoids Mediate Neuron-Astrocyte Communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef]
- Navarrete, M.; Díez, A.; Araque, A. Astrocytes in Endocannabinoid Signalling. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef]
- López, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vázquez, C.; Amores, M.; Ruiz-Pérez, G.; García-García, E.; et al. Cannabinoid CB2 Receptors in the Mouse Brain: Relevance for Alzheimer’s Disease. J. Neuroinflamm. 2018, 15, 158. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Möller, T.; Stella, N. Astrocytes in Culture Produce Anandamide and Other Acylethanolamides. J. Biol. Chem. 2002, 277, 20869–20876. [Google Scholar] [CrossRef]
- Hu, M.; Zhu, D.; Zhang, J.; Gao, F.; Hashem, J.; Kingsley, P.; Marnett, L.J.; MacKie, K.; Chen, C. Enhancing Endocannabinoid Signalling in Astrocytes Promotes Recovery from Traumatic Brain Injury. Brain 2022, 145, 179–193. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef] [PubMed]
- Moshkforoush, A.; Ashenagar, B.; Tsoukias, N.M.; Alevriadou, B.R. Modeling the Role of Endoplasmic Reticulum-Mitochondria Microdomains in Calcium Dynamics. Sci. Rep. 2019, 9, 17072. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E. The Yin and Yang of Mitochondrial Ca2+ Signaling in Cell Physiology and Pathology. Cell Calcium 2021, 93, 102321. [Google Scholar] [CrossRef]
- Sarasija, S.; Norman, K.R. Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis Elegans. Antioxidants 2018, 7, 111. [Google Scholar] [CrossRef]
- Magi, S.; Castaldo, P.; MacRi, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular Calcium Dysregulation: Implications for Alzheimer’s Disease. Biomed. Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (Presenilin 2) Mutants Linked to Familial Alzheimer Disease Impair Autophagy by Altering Ca2+ Homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; VecellioReane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; DeStefani, D.; Rizzuto, R. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef]
- Tufi, R.; Gleeson, T.P.; von Stockum, S.; Hewitt, V.L.; Lee, J.J.; Terriente-Felix, A.; Sanchez-Martinez, A.; Ziviani, E.; Whitworth, A.J. Comprehensive Genetic Characterization of Mitochondrial Ca2+ Uniporter Components Reveals Their Different Physiological Requirements In Vivo. Cell Rep. 2019, 27, 1541–1550.e5. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, A.; Iyer, M.; Balasubramanian, V.; Vellingiri, B. Mitochondrial Calcium Uniporter as a Potential Therapeutic Strategy for Alzheimer’s Disease. Acta Neuropsychiatr. 2020, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased Mitochondrial Calcium Levels Associated with Neuronal Death in a Mouse Model of Alzheimer’s Disease. Nat. Commun 2020, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a Multi-Functional Mitochondrial Protein Regulating Cell Life and Death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 Selectively Transfers Apoptotic Ca2+ Signals to Mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; Van Berlo, J.H.; et al. The Mitochondrial Na+/Ca2+ Exchanger Is Essential for Ca2+ Homeostasis and Viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired Mitochondrial Calcium Efflux Contributes to Disease Progression in Models of Alzheimer’s Disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Andrews, S.B. Calcium-Dependent Mitochondrial Function and Dysfunction in Neurons. FEBS J. 2010, 277, 3622. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (Neurode)Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef]
- Lewis, T.L.; Kwon, S.K.; Lee, A.; Shaw, R.; Polleux, F. MFF-Dependent Mitochondrial Fission Regulates Presynaptic Release and Axon Branching by Limiting Axonal Mitochondria Size. Nat. Commun. 2018, 9, 5008. [Google Scholar] [CrossRef]
- Yin, J.; Reiman, E.M.; Beach, T.G.; Serrano, G.E.; Sabbagh, M.N.; Nielsen, M.; Caselli, R.J.; Shi, J. Effect of ApoE Isoforms on Mitochondria in Alzheimer Disease. Neurology 2020, 94, E2404–E2411. [Google Scholar] [CrossRef]
- Peck, K.J.; Girard, T.A.; Russo, F.A.; Fiocco, A.J. Music and Memory in Alzheimer’s Disease and The Potential Underlying Mechanisms. J. Alzheimers Dis. 2016, 51, 949–959. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional Brain Abnormalities in Young Adults at Genetic Risk for Late-Onset Alzheimer’s Dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, Å.; Aspenstrom, P.; Rizzuto, R.; Hajnóczky, G. Bidirectional Ca2+-Dependent Control of Mitochondrial Dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef]
- Chang, K.T.; Niescier, R.F.; Min, K.T. Mitochondrial Matrix Ca2+ as an Intrinsic Signal Regulating Mitochondrial Motility in Axons. Proc. Natl. Acad. Sci. USA 2011, 108, 15456–15461. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial Abnormalities in Parkinson’s Disease and Alzheimer’s Disease: Can Mitochondria Be Targeted Therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca(2+) Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M.F. The Release of Calcium from the Endoplasmic Reticulum Induced by Amyloid-Beta and Prion Peptides Activates the Mitochondrial Apoptotic Pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef]
- Jia, K.; Du, H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer’s Disease. Cells 2021, 10, 649. [Google Scholar] [CrossRef]
- Krols, M.; Bultynck, G.; Janssens, S. ER-Mitochondria Contact Sites: A New Regulator of Cellular Calcium Flux Comes into Play. J. Cell Biol. 2016, 214, 367–370. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition Pore: Molecular Nature and Role as a Target in Cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer´s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; M McKhann, G.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation and Ameliorates Learning and Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial Dysfunctions in Neurodegenerative Diseases: Relevance to Alzheimer’s Disease. Biomed. Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of Protein Aggregation in Mitochondrial Dysfunction and Neurodegeneration in Alzheimer’s and Parkinson’s Diseases. Neuromol. Med. 2003, 4, 21–35. [Google Scholar] [CrossRef]
- Pérez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Permeability Transition Pore Contributes to Mitochondrial Dysfunction in Fibroblasts of Patients with Sporadic Alzheimer’s Disease. Redox. Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-Associated Membranes (MAMs) and Inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef]
- Yu, W.; Jin, H.; Huang, Y. Mitochondria-Associated Membranes (MAMs): A Potential Therapeutic Target for Treating Alzheimer’s Disease. Clin. Sci. 2021, 135, 109–126. [Google Scholar] [CrossRef]
- Gibson, G.E.; Thakkar, A. Interactions of Mitochondria/Metabolism and Calcium Regulation in Alzheimer’s Disease: A Calcinist Point of View. Neurochem. Res. 2017, 42, 1636–1648. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Kharitonova, E.K.; Bacskai, B.J. Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells 2020, 9, 2513. [Google Scholar] [CrossRef]
- Britzolaki, A.; Saurine, J.; Klocke, B.; Pitychoutis, P.M. A Role for SERCA Pumps in the Neurobiology of Neuropsychiatric and Neurodegenerative Disorders. Adv. Exp. Med. Biol. 2020, 1131, 131–161. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA Pump Activity Is Physiologically Regulated by Presenilin and Regulates Amyloid Beta Production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M.F. Involvement of Endoplasmic Reticulum Ca2+ Release through Ryanodine and Inositol 1,4,5-Triphosphate Receptors in the Neurotoxic Effects Induced by the Amyloid-Beta Peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef]
- Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 9, 2577. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s Disease-Linked Presenilin Mutants and Intracellular Ca2+ Handling: A Single-Organelle, FRET-Based Analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, S. Impairment of Store-Operated Calcium Entry: Implications in Alzheimer’s Neurodegeneration. Curr. Alzheimer Res. 2020, 17, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Forms and Functions of Store-Operated Calcium Entry Mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Pomorski, P.; Wisniewska, M.B.; Kuznicki, J. Differential Roles for STIM1 and STIM2 in Store-Operated Calcium Entry in Rat Neurons. PLoS ONE 2011, 6, e19285. [Google Scholar] [CrossRef] [PubMed]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’angelo, E. STIM and Orai Proteins in Neuronal Ca2+ Signaling and Excitability. Front. Cell Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Scremin, E.; Agostini, M.; Leparulo, A.; Pozzan, T.; Greotti, E.; Fasolato, C. ORAI2 Down-Regulation Potentiates SOCE and Decreases Aβ42 Accumulation in Human Neuroglioma Cells. Int. J. Mol. Sci. 2020, 21, 5288. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron Loss in the 5XFAD Mouse Model of Alzheimer’s Disease Correlates with Intraneuronal Aβ42 Accumulation and Caspase-3 Activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar] [CrossRef]
- Bruno, A.M.; Huang, J.Y.; Bennett, D.A.; Marr, R.A.; Hastings, M.L.; Stutzmann, G.E. Altered Ryanodine Receptor Expression in Mild Cognitive Impairment and Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1001.e1–1001.e6. [Google Scholar] [CrossRef]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant Ryanodine Receptor-Mediated Calcium Release Resets Synaptic Homeostasis in Presymptomatic 3xTg-AD Mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Liu, J.; Supnet, C.; Sun, S.; Zhang, H.; Good, L.; Popugaeva, E.; Bezprozvanny, I. The Role of Ryanodine Receptor Type 3 in a Mouse Model of Alzheimer Disease. Channels 2014, 8, 230–242. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Wei, H. Dantrolene: From Malignant Hyperthermia to Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 668–676. [Google Scholar] [CrossRef]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine Receptor Blockade Reduces Amyloid-β Load and Memory Impairments in Tg2576 Mouse Model of Alzheimer Disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef]
- Fruen, B.R.; Mickelson, J.R.; Louis, C.F. Dantrolene Inhibition of Sarcoplasmic Reticulum Ca2+ Release by Direct and Specific Action at Skeletal Muscle Ryanodine Receptors. J. Biol. Chem. 1997, 272, 26965–26971. [Google Scholar] [CrossRef]
- Veeresh, P.; Kaur, H.; Sarmah, D.; Mounica, L.; Verma, G.; Kotian, V.; Kesharwani, R.; Kalia, K.; Borah, A.; Wang, X.; et al. Endoplasmic Reticulum-Mitochondria Crosstalk: From Junction to Function across Neurological Disorders. Ann. N. Y. Acad. Sci. 2019, 1457, 41–60. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimers Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; De Strooper, B.; Bezprozvanny, I. Familial Alzheimer Disease-Linked Mutations Specifically Disrupt Ca2+ Leak Function of Presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin Mutations Linked to Familial Alzheimer’s Disease Reduce Endoplasmic Reticulum and Golgi Apparatus Calcium Levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced Expression of the Voltage-Dependent Anion Channel 1 (VDAC1) in Alzheimer’s Disease Transgenic Mice: An Insight into the Pathogenic Effects of Amyloid-β. J. Alzheimers Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the Endoplasmic Reticulum-Mitochondria Interface in Alzheimer’s Disease and Related Models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I. Cytotoxicity of Intracellular Aβ42 Amyloid Oligomers Involves Ca2+ Release from the Endoplasmic Reticulum by Stimulated Production of Inositol Trisphosphate. J. Neurosci. 2013, 33, 3824–3833. [Google Scholar] [CrossRef]
- Shilling, D.; Müller, M.; Takano, H.; Mak, D.O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 Receptor-Mediated Ca2+ Signaling Alleviates Mutant Presenilin-Linked Familial Alzheimer’s Disease Pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 Upregulates the Activity of Mitochondria-Associated ER Membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Iwata, N. Memantine Monotherapy for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0123289. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. A Novel NMDA Receptor Antagonist Protects against Cognitive Decline Presented by Senescent Mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ Induces Astrocytic Glutamate Release, Extrasynaptic NMDA Receptor Activation, and Synaptic Loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef]
- Michalska, P.; Mayo, P.; Fernández-Mendívil, C.; Tenti, G.; Duarte, P.; Buendia, I.; Ramos, M.T.; López, M.G.; Menéndez, J.C.; León, R. Antioxidant, Anti-Inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease. Antioxidants 2020, 9, 650. [Google Scholar] [CrossRef]
- Ismaili, L.; Monnin, J.; Etievant, A.; Arribas, R.L.; Viejo, L.; Refouvelet, B.; Soukup, O.; Janockova, J.; Hepnarova, V.; Korabecny, J.; et al. (±)- BIGI-3h: Pentatarget-Directed Ligand Combining Cholinesterase, Monoamine Oxidase, and Glycogen Synthase Kinase 3β Inhibition with Calcium Channel Antagonism and Antiaggregating Properties for Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1328–1342. [Google Scholar] [CrossRef]
- Ponne, S.; Kumar, C.R.; Boopathy, R. Verapamil Attenuates Scopolamine Induced Cognitive Deficits by Averting Oxidative Stress and Mitochondrial Injury—A Potential Therapeutic Agent for Alzheimer’s Disease. Metab. Brain Dis. 2020, 35, 503–515. [Google Scholar] [CrossRef]
- Bai, J.Z.; Lipski, J. Involvement of TRPV4 Channels in Aβ40-Induced Hippocampal Cell Death and Astrocytic Ca2+ Signalling. Neurotoxicology 2014, 41, 64–72. [Google Scholar] [CrossRef]
- Deng, Y.; Li, W.; Niu, L.; Luo, X.; Li, J.; Zhang, Y.; Liu, H.; He, J.; Wan, W. Amelioration of Scopolamine-Induced Learning and Memory Impairment by the TRPV4 Inhibitor HC067047 in ICR Mice. Neurosci. Lett. 2022, 767, 136209. [Google Scholar] [CrossRef]
- Storozhuk, M.V.; Moroz, O.F.; Zholos, A.V. Multifunctional TRPV1 Ion Channels in Physiology and Pathology with Focus on the Brain, Vasculature, and Some Visceral Systems. Biomed. Res. Int. 2019, 2019, 5806321. [Google Scholar] [CrossRef]
- Jung, K.M.; Astarita, G.; Yasar, S.; Vasilevko, V.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. An Amyloid Β42-Dependent Deficit in Anandamide Mobilization Is Associated with Cognitive Dysfunction in Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1522–1532. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. Cannabinoids for Treatment of Alzheimer’s Disease: Moving toward the Clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Balleza-Tapia, H.; Crux, S.; Andrade-Talavera, Y.; Dolz-Gaiton, P.; Papadia, D.; Chen, G.; Johansson, J.; Fisahn, A. TrpV1 Receptor Activation Rescues Neuronal Function and Network Gamma Oscillations from Aβ-Induced Impairment in Mouse Hippocampus in Vitro. Elife 2018, 7, e37703. [Google Scholar] [CrossRef]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef]
- Chen, M.L.; Hong, C.G.; Yue, T.; Li, H.M.; Duan, R.; Hu, W.B.; Cao, J.; Wang, Z.X.; Chen, C.Y.; Hu, X.K.; et al. Inhibition of MiR-331-3p and MiR-9-5p Ameliorates Alzheimer’s Disease by Enhancing Autophagy. Theranostics 2021, 11, 2395–2409. [Google Scholar] [CrossRef]
- Wang, C.; Huang, W.; Lu, J.; Chen, H.; Yu, Z. TRPV1-Mediated Microglial Autophagy Attenuates Alzheimer’s Disease-Associated Pathology and Cognitive Decline. Front. Pharmacol. 2022, 12, 3809. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Kim, J.; Ham, S.; Park, J.H.Y.; Han, S.; Jung, Y.K.; Shim, I.; Han, J.S.; Lee, K.W.; et al. Ca2+-Permeable TRPV1 Pain Receptor Knockout Rescues Memory Deficits and Reduces Amyloid-β and Tau in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2020, 29, 228–237. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Tacer, K.F.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef]
- Popugaeva, E.; Chernyuk, D.; Zhang, H.; Postnikova, T.Y.; Pats, K.; Fedorova, E.; Poroikov, V.; Zaitsev, A.V.; Bezprozvanny, I. Derivatives of Piperazines as Potential Therapeutic Agents for Alzheimer’s Disease. Mol. Pharmacol. 2019, 95, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, R.; Yang, J.; Li, H.; He, Z.; Jing, N.; Wang, X.; Wang, Y. TRPC6 Specifically Interacts with APP to Inhibit Its Cleavage by γ-Secretase and Reduce Aβ Production. Nat. Commun. 2015, 6, 8876. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Lu, R.; Wang, J.; Zeng, S.; Zhang, T.; Guo, W.; Zhang, X.; Cheng, Q.; Yue, C.; Wang, Y.; et al. Probing the Therapeutic Potential of TRPC6 for Alzheimer’s Disease in Live Neurons from Patient-Specific IPSCs. J. Mol. Cell Biol. 2020, 12, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Montecinos-Oliva, C.; Schüller, A.; Inestrosa, N.C. Tetrahydrohyperforin: A Neuroprotective Modified Natural Compound against Alzheimer’s Disease. Neural Regen. Res. 2015, 10, 552–554. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Tapia-Rojas, C.; Griffith, T.N.; Carvajal, F.J.; Benito, M.J.; Rivera-Dictter, A.; Alvarez, A.R.; Serrano, F.G.; Hancke, J.L.; Burgos, P.V.; et al. Tetrahydrohyperforin Prevents Cognitive Deficit, Aβ Deposition, Tau Phosphorylation and Synaptotoxicity in the APPswe/PSEN1ΔE9 Model of Alzheimer’s Disease: A Possible Effect on APP Processing. Transl. Psychiatry 2011, 1, e20. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Zolezzi, J.M.; Tapia-Rojas, C.; Godoy, J.A.; Inestrosa, N.C. Tetrahydrohyperforin Decreases Cholinergic Markers Associated with Amyloid-β Plaques, 4-Hydroxynonenal Formation, and Caspase-3 Activation in AβPP/PS1 Mice. J. Alzheimers Dis. 2013, 36, 99–118. [Google Scholar] [CrossRef]
- Cerpa, W.; Hancke, J.; Morazzoni, P.; Bombardelli, E.; Riva, A.; Marin, P.; Inestrosa, N. The Hyperforin Derivative IDN5706 Occludes Spatial Memory Impairments and Neuropathological Changes in a Double Transgenic Alzheimer’s Mouse Model. Curr. Alzheimer Res. 2010, 7, 126–133. [Google Scholar] [CrossRef]
- Liu, L.; Chen, M.; Lin, K.; Xiang, X.; Yang, J.; Zheng, Y.; Xiong, X.; Zhu, S. TRPC6 Attenuates Cortical Astrocytic Apoptosis and Inflammation in Cerebral Ischemic/Reperfusion Injury. Front. Cell Dev. Biol. 2021, 8, 594283. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Xu, H.; Zeng, Z.; Zhu, H. TRPML1 Participates in the Progression of Alzheimer’s Disease by Regulating the PPARγ/AMPK/Mtor Signalling Pathway. Cell. Physiol. Biochem. 2017, 43, 2446–2456. [Google Scholar] [CrossRef]
- Somogyi, A.; Kirkham, E.D.; Lloyd-Evans, E.; Allen, N.D.; Anderson, K.E.; Hawkins, P.T.; Sims, R.; Boland, B.; O’Neill, C. TRPML1: A Novel Therapeutic Target to Remediate Endolysosomal Pathology in Alzheimer’s Disease. Alzheimer’s Dement. 2020, 16, e043397. [Google Scholar] [CrossRef]
- Granatiero, V.; Pacifici, M.; Raffaello, A.; De Stefani, D.; Rizzuto, R. Overexpression of Mitochondrial Calcium Uniporter Causes Neuronal Death. Oxid. Med. Cell Longev. 2019, 2019, 1681254. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Tan, Y.W.; Hagenston, A.M.; Martel, M.A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.A.; Bading, H.; Hardingham, G.E. Mitochondrial Calcium Uniporter Mcu Controls Excitotoxicity and Is Transcriptionally Repressed by Neuroprotective Nuclear Calcium Signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial Dysfunction Is a Trigger of Alzheimer’s Disease Pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Liu, X.; Xun, Q.; Zhang, X. Neuroprotective Effect of Ginkgolide K against H2O2-Induced PC12 Cell Cytotoxicity by Ameliorating Mitochondrial Dysfunction and Oxidative Stress. Biol. Pharm. Bull. 2014, 37, 217–225. [Google Scholar] [CrossRef]
- Liu, H.; Li, Q.; Zhang, X.; Shi, Y.; Li, J. Effect of Ginkgolide K on Calcium Channel Activity in Alzheimer’s Disease. Exp. Ther. Med. 2022, 23, 426. [Google Scholar] [CrossRef]
- Berrocal, M.; Corbacho, I.; Vázquez-Hernández, M.; Ávila, J.; Sepúlveda, M.R.; Mata, A.M. Inhibition of PMCA Activity by Tau as a Function of Aging and Alzheimer’s Neuropathology. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1465–1476. [Google Scholar] [CrossRef]
- Berrocal, M.; Saez, L.; Mata, A.M. Sorcin Activates the Brain Pmca and Blocks the Inhibitory Effects of Molecular Markers of Alzheimer’s Disease on the Pump Activity. Int. J. Mol. Sci. 2021, 22, 6055. [Google Scholar] [CrossRef]
- Berrocal, M.; Corbacho, I.; Gutierrez-Merino, C.; Mata, A.M. Methylene Blue Activates the PMCA Activity and Cross-Interacts with Amyloid β-Peptide, Blocking Aβ-Mediated PMCA Inhibition. Neuropharmacology 2018, 139, 163–172. [Google Scholar] [CrossRef]
- Afewerky, H.K.; Li, H.; Pang, P.; Zhang, T.; Lu, Y. Contribution of Sodium-Calcium Exchanger Isoform-3 in Aβ1-42 Induced Cell Death. Neuropsychiatry 2019, 9, 2220–2227. [Google Scholar] [CrossRef]
- Afewerky, H.K.; Li, H.; Zhang, T.; Li, X.; Mahaman, Y.A.R.; Duan, L.; Qin, P.; Zheng, J.; Pei, L.; Lu, Y. Sodium–Calcium Exchanger Isoform-3 Targeted Withania somnifera (L.) Dunal Therapeutic Intervention Ameliorates Cognition in the 5xFAD Mouse Model of Alzheimer’s Disease. Sci. Rep. 2022, 12, 1537. [Google Scholar] [CrossRef]
- Saykally, J.N.; Hatic, H.; Keeley, K.L.; Jain, S.C.; Ravindranath, V.; Citron, B.A. Withania somnifera Extract Protects Model Neurons from In Vitro Traumatic Injury. Cell Transplant. 2017, 26, 1193–1201. [Google Scholar] [CrossRef]
- Ryazantseva, M.; Goncharova, A.; Skobeleva, K.; Erokhin, M.; Methner, A.; Georgiev, P.; Kaznacheyeva, E. Presenilin-1 Delta E9 Mutant Induces STIM1-Driven Store-Operated Calcium Channel Hyperactivation in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 4667–4680. [Google Scholar] [CrossRef]
- Chernyuk, D.; Zernov, N.; Kabirova, M.; Bezprozvanny, I.; Popugaeva, E. Antagonist of Neuronal Store-Operated Calcium Entry Exerts Beneficial Effects in Neurons Expressing PSEN1ΔE9 Mutant Linked to Familial Alzheimer Disease. Neuroscience 2019, 410, 118–127. [Google Scholar] [CrossRef]
- Pohanka, M. Alpha7 Nicotinic Acetylcholine Receptor Is a Target in Pharmacology and Toxicology. Int. J. Mol. Sci. 2012, 13, 2219–2238. [Google Scholar] [CrossRef]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 Nicotinic Acetylcholine Receptor-Specific Agonist DMXBA (GTS-21) Attenuates Aβ Accumulation through Suppression of Neuronal γ-Secretase Activity and Promotion of Microglial Amyloid-β Phagocytosis and Ameliorates Cognitive Impairment in a Mouse Mode. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar] [CrossRef]
- Florian, H.; Meier, A.; Gauthier, S.; Lipschitz, S.; Lin, Y.; Tang, Q.; Othman, A.A.; Robieson, W.Z.; Gault, L.M. Efficacy and Safety of ABT-126 in Subjects with Mild-to-Moderate Alzheimer’s Disease on Stable Doses of Acetylcholinesterase Inhibitors: A Randomized, Double-Blind, Placebo-Controlled Study. J. Alzheimers Dis. 2016, 51, 1237–1247. [Google Scholar] [CrossRef]
- Potasiewicz, A.; Krawczyk, M.; Gzielo, K.; Popik, P.; Nikiforuk, A. Positive Allosteric Modulators of Alpha 7 Nicotinic Acetylcholine Receptors Enhance Procognitive Effects of Conventional Anti-Alzheimer Drugs in Scopolamine-Treated Rats. Behav. Brain Res. 2020, 385, 112547. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Gao, X.; Zhang, J.; Ben Abou, M.; Liang, G.; Meng, Q.; Hepner, A.; Eckenhoff, M.F.; Wei, H.; et al. Intranasal Dantrolene as a Disease-Modifying Drug in Alzheimer 5XFAD Mice. J. Alzheimers Dis. 2020, 76, 1375–1389. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, G.; Liang, S.; Mund, R.; Shi, Y.; Wei, H. Dantrolene Ameliorates Impaired Neurogenesis and Synaptogenesis in Induced Pluripotent Stem Cell Lines Derived from Patients with Alzheimer’s Disease. Anesthesiology 2020, 132, 1062–1079. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, J.; Song, Z.; Guo, W.; Sun, B.; Wei, J.; Estillore, J.P.; Back, T.G.; Chen, S.R.W. Limiting RyR2 Open Time Prevents Alzheimer’s Disease-Related Deficits in the 3xTG-AD Mouse Model. J. Neurosci. Res. 2021, 99, 2906–2921. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ Disruption in Alzheimer’s Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Zhao, F.; Wang, C.F.; Zhang, X.M.; Xiao, Y.; Zhou, F.; Wu, M.N.; Zhang, J.; Qi, J.S.; Yang, W.; et al. Xestospongin C, a Reversible IP 3 Receptor Antagonist, Alleviates the Cognitive and Pathological Impairments in APP/PS1 Mice of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Sander, P.; Gudermann, T.; Schredelseker, J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? Int. J. Mol. Sci. 2021, 22, 946. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal Interaction of VDAC1 with Amyloid Beta and Phosphorylated Tau Causes Mitochondrial Dysfunction in Alzheimer’s Disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Wang, D.M.; Li, S.Q.; Zhu, X.Y.; Wang, Y.; Wu, W.L.; Zhang, X.J. Protective Effects of Hesperidin against Amyloid-β (Aβ) Induced Neurotoxicity through the Voltage Dependent Anion Channel 1 (VDAC1)-Mediated Mitochondrial Apoptotic Pathway in PC12 Cells. Neurochem. Res. 2013, 38, 1034–1044. [Google Scholar] [CrossRef]
- Maccarrone, M.; Totaro, A.; Leuti, A.; Giacovazzo, G.; Scipioni, L.; Mango, D.; Coccurello, R.; Nisticò, R.; Oddi, S. Early Alteration of Distribution and Activity of Hippocampal Type-1 Cannabinoid Receptor in Alzheimer’s Disease-like Mice Overexpressing the Human Mutant Amyloid Precursor Protein. Pharmacol. Res. 2018, 130, 366–373. [Google Scholar] [CrossRef]
- Aguirre-Rueda, D.; Guerra-Ojeda, S.; Aldasoro, M.; Iradi, A.; Obrador, E.; Mauricio, M.D.; Vila, J.M.; Marchio, P.; Valles, S.L. WIN 55,212-2, Agonist of Cannabinoid Receptors, Prevents Amyloid Β1-42 Effects on Astrocytes in Primary Culture. PLoS ONE 2015, 10, e0122843. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; de Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol Reduces Aβ-Induced Neuroinflammation and Promotes Hippocampal Neurogenesis through PPARγ Involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in Vivo Blunts Beta-Amyloid Induced Neuroinflammation by Suppressing IL-1beta and INOS Expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, P.; Xie, Y.; Chen, X.; Solowij, N.; Green, K.; Chew, Y.L.; Huang, X.F. Cannabidiol Regulates CB1-PSTAT3 Signaling for Neurite Outgrowth, Prolongs Lifespan, and Improves Health Span in Caenorhabditis Elegans of Aβ Pathology Models. FASEB J. 2021, 35, e21537. [Google Scholar] [CrossRef]
- Yamagata, Y.; Kobayashi, S.; Umeda, T.; Inoue, A.; Sakagami, H.; Fukaya, M.; Watanabe, M.; Hatanaka, N.; Totsuka, M.; Yagi, T.; et al. Kinase-Dead Knock-In Mouse Reveals an Essential Role of Kinase Activity of Ca2+/Calmodulin-Dependent Protein Kinase IIα in Dendritic Spine Enlargement, Long-Term Potentiation, and Learning. J. Neurosci. 2009, 29, 7607–7618. [Google Scholar] [CrossRef]
- O’day, D.H. Calmodulin Binding Proteins and Alzheimer’s Disease: Biomarkers, Regulatory Enzymes and Receptors That Are Regulated by Calmodulin. Int. J. Mol. Sci. 2020, 21, 7344. [Google Scholar] [CrossRef]
- Chen, C.; Xu, D.; Zhang, Z.H.; Jia, S.Z.; Cao, X.C.; Chen, Y.B.; Song, G.L.; Wong, M.S.; Li, H.W. Cognitive Improvement and Synaptic Deficit Attenuation by a Multifunctional Carbazole-Based Cyanine in AD Mice Model through Regulation of Ca2+/CaMKII/CREB Signaling Pathway. Exp. Neurol. 2020, 327, 113210. [Google Scholar] [CrossRef]
- Al Rihani, S.B.; Lan, R.S.; Kaddoumi, A.; Gosselet, F. Granisetron Alleviates Alzheimer’s Disease Pathology in TgSwDI Mice Through Calmodulin-Dependent Protein Kinase II/CAMP-Response Element Binding Protein Pathway. J. Alzheimers Dis. 2019, 72, 1097–1117. [Google Scholar] [CrossRef]
- Hudry, E.; Wu, H.Y.; Arbel-Ornath, M.; Hashimoto, T.; Matsouaka, R.; Fan, Z.; Spires-Jones, T.L.; Betensky, R.A.; Bacskai, B.J.; Hyman, B.T. Inhibition of the NFAT Pathway Alleviates Amyloid Beta Neurotoxicity in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2012, 32, 3176–3192. [Google Scholar] [CrossRef]
- Rozkalne, A.; Hyman, B.T.; Spires-Jones, T.L. Calcineurin Inhibition with FK506 Ameliorates Dendritic Spine Density Deficits in Plaque-Bearing Alzheimer Model Mice. Neurobiol. Dis. 2011, 41, 650–654. [Google Scholar] [CrossRef]
- Köylü, A.; Altunkaynak, B.Z.; Delibaş, B. Effects of Tacrolimus on C-Fos in Hippocampus and Memory Performances in Streptozotocin Model of Alzheimer’s Disease of Rats. Turk. J. Med. Sci. 2021, 51, 2159–2166. [Google Scholar] [CrossRef]
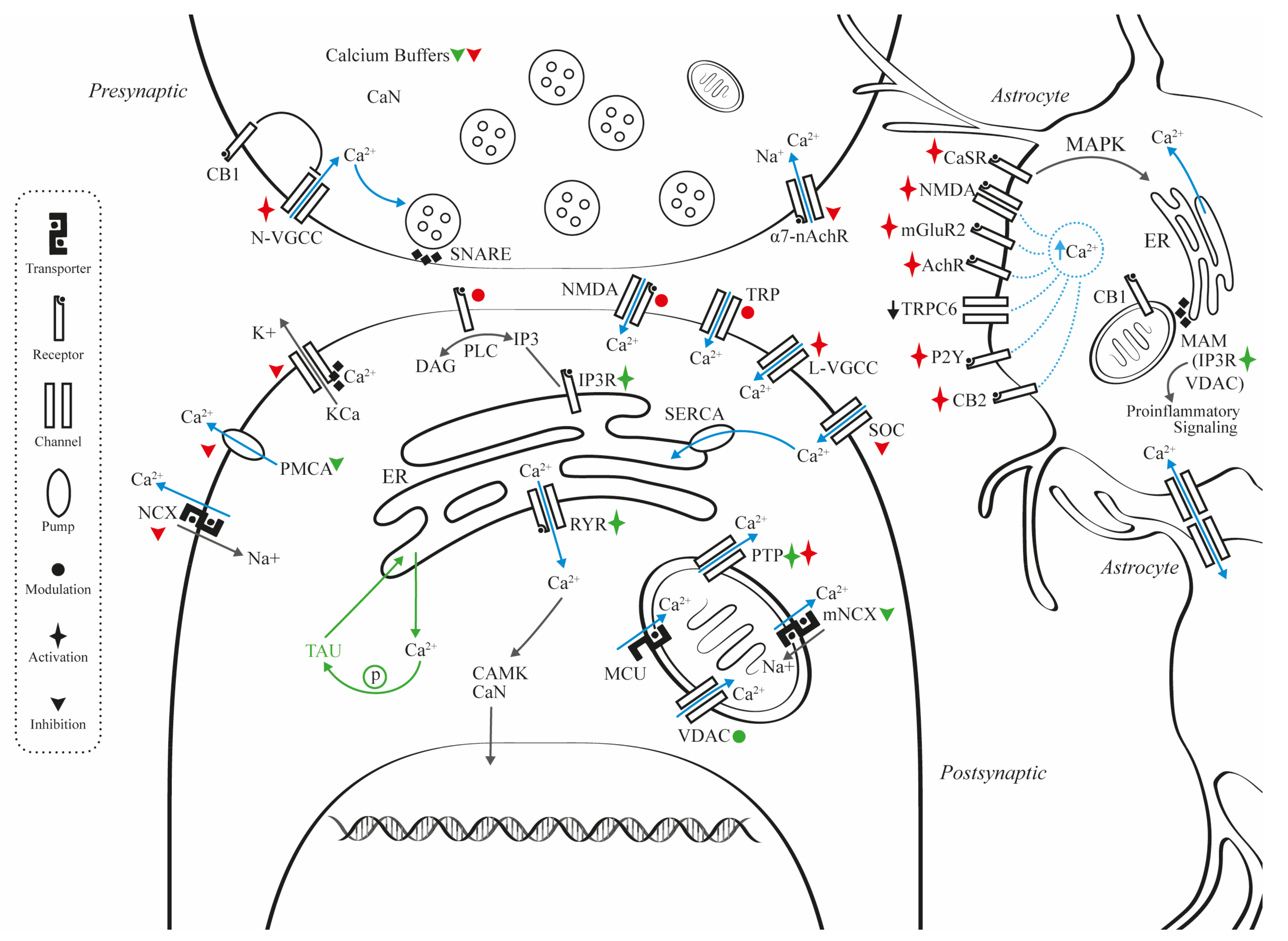
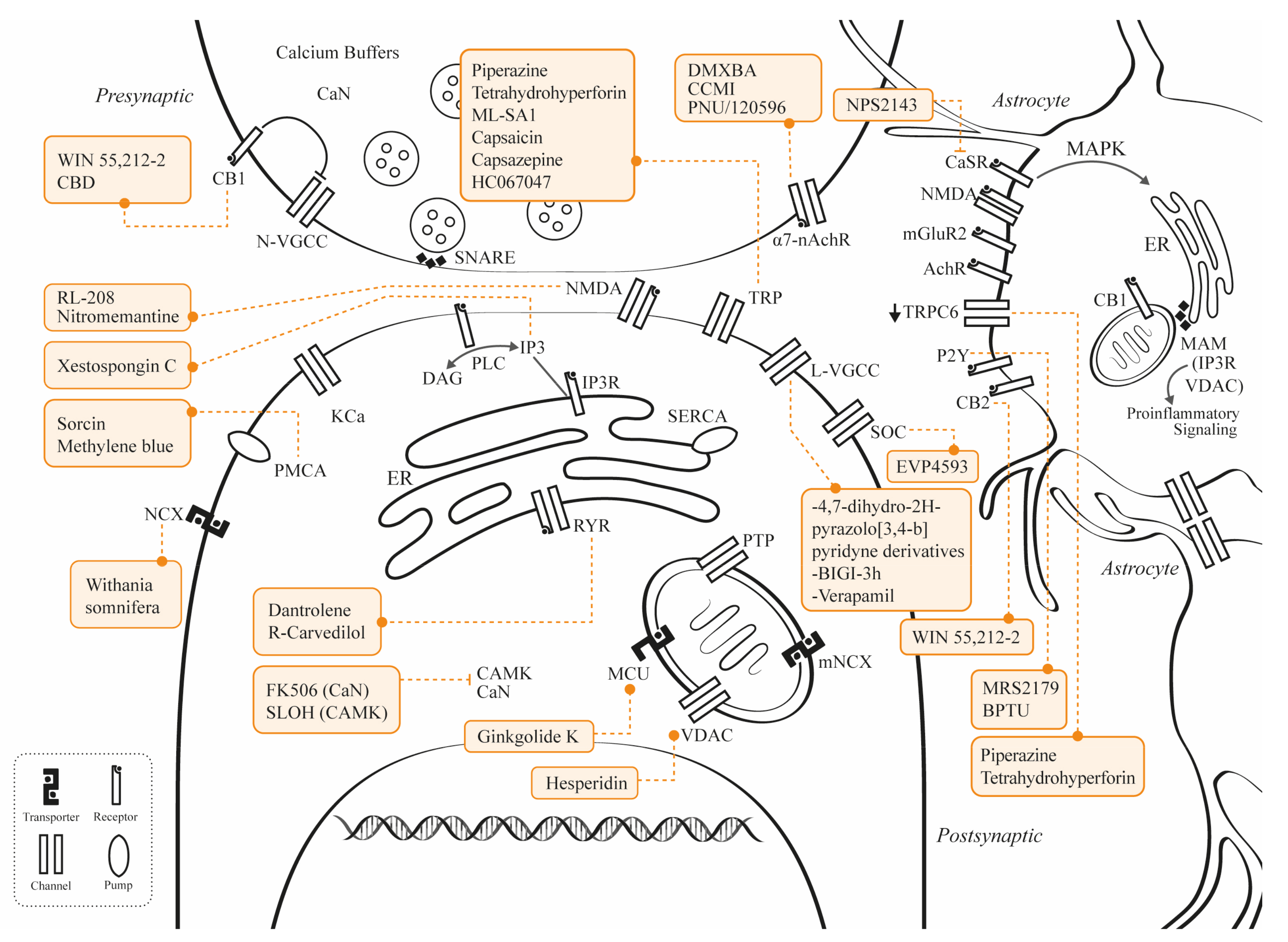

{kind=link}
{kind=link}
| Molecular Target | Therapeutic Agent | Model | Therapeutic Effect | Reference |
|---|---|---|---|---|
| α7nAChR | DMXBA | Primary cultures of rat microglia and transgenic mouse model (APdE9 mice) | Increased Aβ phagocytosis | [337] |
| CCMI PNU-120596 | Scopolamine-induced memory deficits in rats | When used in combination with AD-approved drugs they reversed the scopolamine-induced deficit | [339] | |
| CaN | FK506 | APP/PS mice | Short-term treatment resulted in amelioration of dendritic spine loss | [358] |
| FK506 | APP-KI mice | Reduced LTP impairment in APP-KI mice | [157] | |
| CaMKII | SLOH | 3xTG-AD | Attenuated synaptic deficit in vivo by regulating the calcium/CaMKII/CREB-signaling pathway Reduced Aβ deposition | [355] |
| CaSR | NPS2143 | Human cortical astrocytes and HCN-1A neurons | Attenuated Aβ1–42 secretion Reduced total CaSR protein complement Blocked excess NO production in human astrocytes Reduced the secretion of IL-6, ICAM-1 | [202,203,204] |
| CB1 | WIN 55,212-2 | Primary cultured rat astrocytes treated with Aβ1–42 | Increased cell viability and anti-inflammatory response | [349] |
| CBD | Mice inoculated with human Aβ1–42 | Reduced GFAP overexpression Reduced expression of iNOS and IL-1β | [350] | |
| CBD | SH-SY5Y neuronal cells | Protected the reduction in dendritic spine density, increased the expression of CB1 receptor, and prevented neurite lesions from Aβ1–42 | [352] | |
| IP3 | Xestospongin C | APP/PS1 mice | Improved cognitive behavior, reduced the number of Aβ plaques, and reduced the expression or ER stress proteins | [344] |
| L-type voltage-dependent calcium antagonist | 4,7-dihydro-2H-pyrazolo[3,4-b] pyridine derivatives | SK-N-SH neuroblastoma cell line exposed to okadaic acid, rotetone, and K+ | Improved cell viability | [299] |
| Rat hippocampal slices treated with okaic acid | Reduced cellular death Reduced oxidative stress | [299] | ||
| BIGI-3h | Scopolamine-induced cognitive dysfunction in mice evaluated with the NORT | Mice treated with BIGI-3h after scopolamine treatment showed significantly improved recognition index | [300] | |
| Verapamil | Human neuroblastoma cells treated with scopolamine | Attenuated the downregulation of mACR1, GAP43, SYP, CREB1, CREBBP, and BDNF | [301] | |
| Scopolamine-treated mice | Attenuated the cognitive and behavioral deficits induced by scopolamine | [301] | ||
| MCU | Ginkgolide K | APP/PS1 mice | Downregulated MCU Improved cognitive ability | [327] |
| NCX3 | Withania somnifera | 5xFAD mice | Significantly improved performance on Barnes circular-maze task Increased NCX3 expression Reduced oxidative stress | [332] |
| NMDA | RL-208 | Senescence-accelerated mice prone 8 (SAMP8), a mouse model of late-onset AD (LOAD). | Improved social behavior and restored cognitive impairment Increased levels of p-NMDAR2B mBDNF Synaptophysin PSD95 | [297] |
| Nitromemantine | 3xTg-AD mice | Improved function on the location-novelty-recognition test | [298] | |
| PMCA | Sorcin | SH-SY5Y Cells | Preservation of PMCA activity Reduced toxicity of SH-SY5Y cells induced by Aβ | [329] |
| Methylene blue | Brain tissues of human patients with AD | Activated PMCA Blocked the inhibitory effect of Aβ on PMCA activity | [330] | |
| P2Y1 | MRS2179 BPTU | APP/PS1 mice | Decreased astrocytic hyperactivity Reversed synaptic deficits | [190] |
| RyR | Dantrolene | 5XFAD mice | Ameliorated memory loss | [340] |
| iPSC from SAD and FAD patients | Increased cell viability and proliferation Restored intracellular calcium homeostasis | [341] | ||
| R-Carvedilol | 3xTg-AD | Rescued memory impairment, LTP deficit, and neuron loss | [342] | |
| nSOCE | EVP4593 | PS1ΔE9-transfected hippocampal neurons | Rescued mushroom-spine loss | [335] |
| TRPC6 | Piperazine | 5xFAD mice | Restored nSOCE in hippocampal neurons Restored LTP in 5xFAD mice | [313] |
| Tetrahydrohyperforin | AβPP/PS transgenic mice | Decreased caspase 3 activation, tau phosphorylation, and Aβ accumulation | [318] | |
| TRPML1 | ML-SA1 | Postmortem LOAD hippocampal neurons | Restored endolysosomal calcium pool to normal levels Decreased endolysosomal swelling Increased the levels of non-pathogenic fragments of APP | [322] |
| TRPV1 | Capsaicin | Rat hippocampal slices treated with recombinant Aβ1–42 | Prevented the reduction in hippocampal gamma oscillations | [307] |
| Capsazepine | 3xTg-AD-derived primary neuronal cultures | Decreased production of Aβ, tau, and p-tau | [311] | |
| Capsaicin | 3xTg mice | Decreased the levels of phosphorylated tau Improved outcomes in NORT and on Y maze Promoted microglial autophagy | [303,310] | |
| TRPV4 | HC067047 | Scopolamine-induced cognitive dysfunction in mice | Reduced scopolamine-induced cognitive dysfunction, as assessed by NPRT and NORT Reduced levels of Bax and caspase 3 Increased levels of NeuN | [303] |
| VDAC1 | Hesperidin | Rat PC12 cells exposed to Aβ25–35 | Reduced Aβ 25–35-induced apoptosis | [347] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; González-Reyes, R.E.; Nava-Mesa, M.O. Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 9067. https://doi.org/10.3390/ijms24109067
Baracaldo-Santamaría D, Avendaño-Lopez SS, Ariza-Salamanca DF, Rodriguez-Giraldo M, Calderon-Ospina CA, González-Reyes RE, Nava-Mesa MO. Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(10):9067. https://doi.org/10.3390/ijms24109067
Chicago/Turabian StyleBaracaldo-Santamaría, Daniela, Sara Sofia Avendaño-Lopez, Daniel Felipe Ariza-Salamanca, Mateo Rodriguez-Giraldo, Carlos A. Calderon-Ospina, Rodrigo E. González-Reyes, and Mauricio O. Nava-Mesa. 2023. "Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 10: 9067. https://doi.org/10.3390/ijms24109067
APA StyleBaracaldo-Santamaría, D., Avendaño-Lopez, S. S., Ariza-Salamanca, D. F., Rodriguez-Giraldo, M., Calderon-Ospina, C. A., González-Reyes, R. E., & Nava-Mesa, M. O. (2023). Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(10), 9067. https://doi.org/10.3390/ijms24109067