

Sequential Treatment with Temozolomide Plus Naturally Derived AT101 as an Alternative Therapeutic Strategy: Insights into Chemoresistance Mechanisms of Surviving Glioblastoma Cells
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
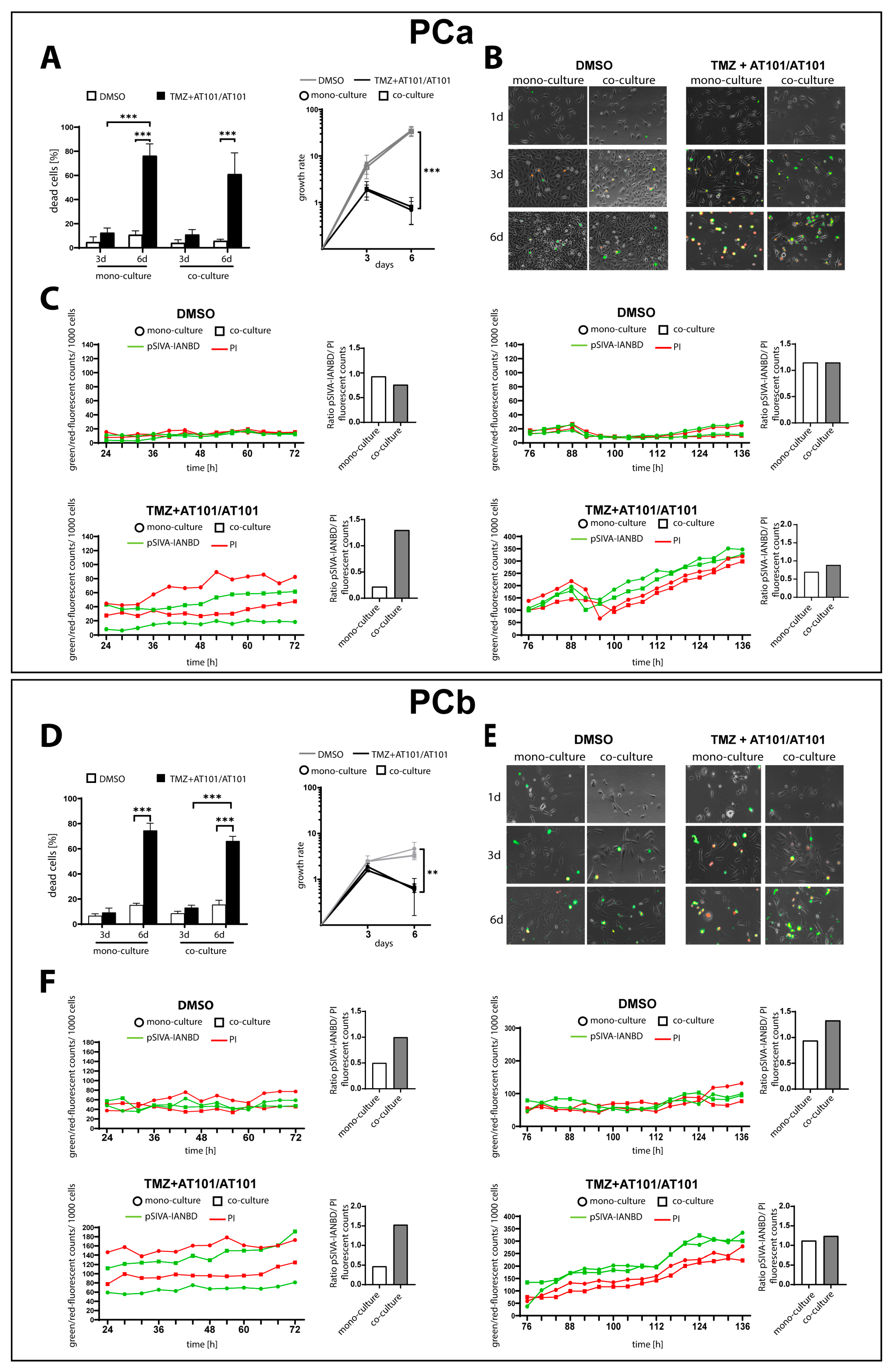
2.1. TMZ+AT101/AT101 Treatment Led to an Increased Number of Surviving Phosphatidylserine-Positive GBM Cells over Time
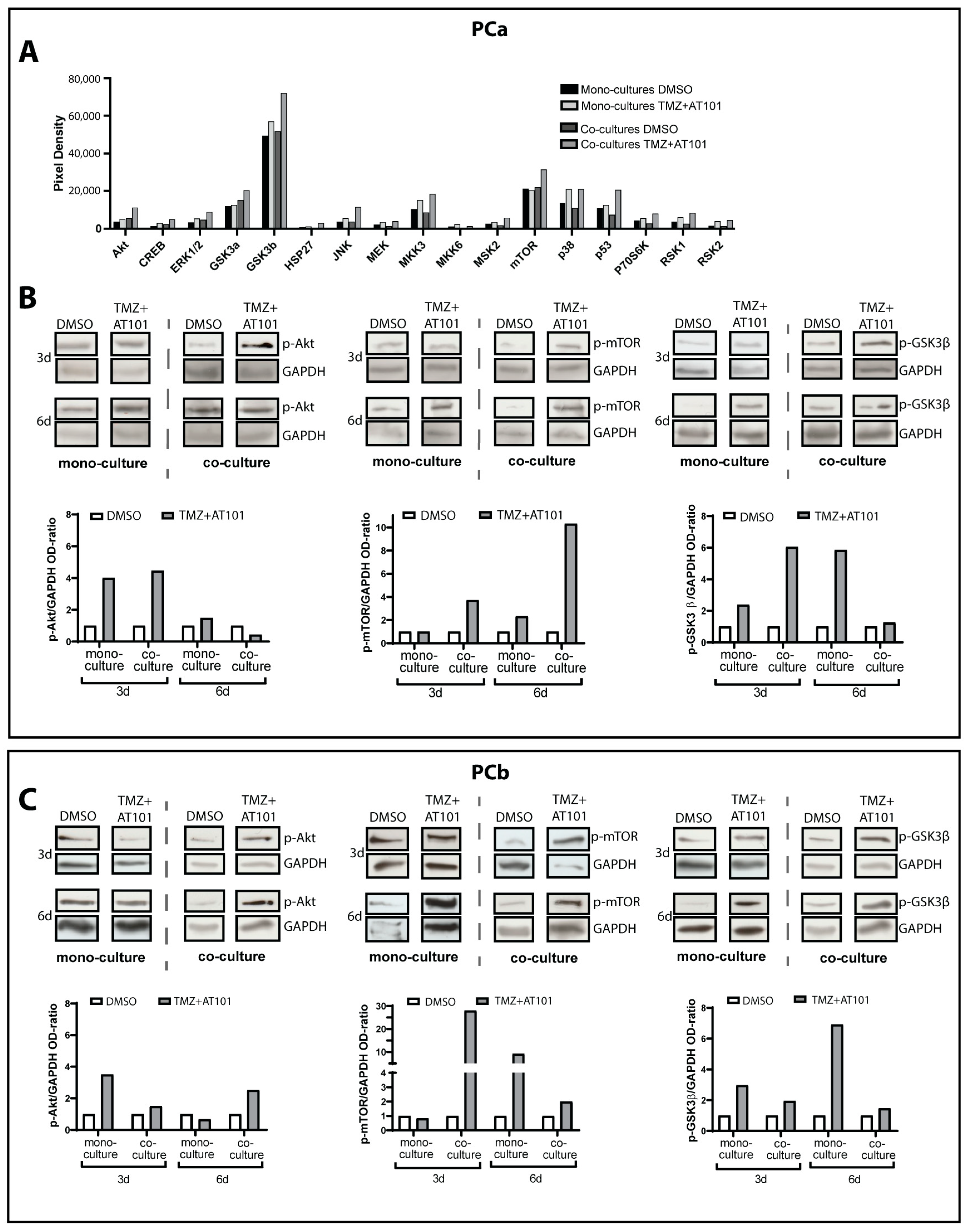
2.2. TMZ+AT101/AT101 Treatment Led to Phosphorylation of AKT, mTOR and GSK3ß, Resulting in Expression of Pro-Tumorigenic Genes in Surviving GBM Cells
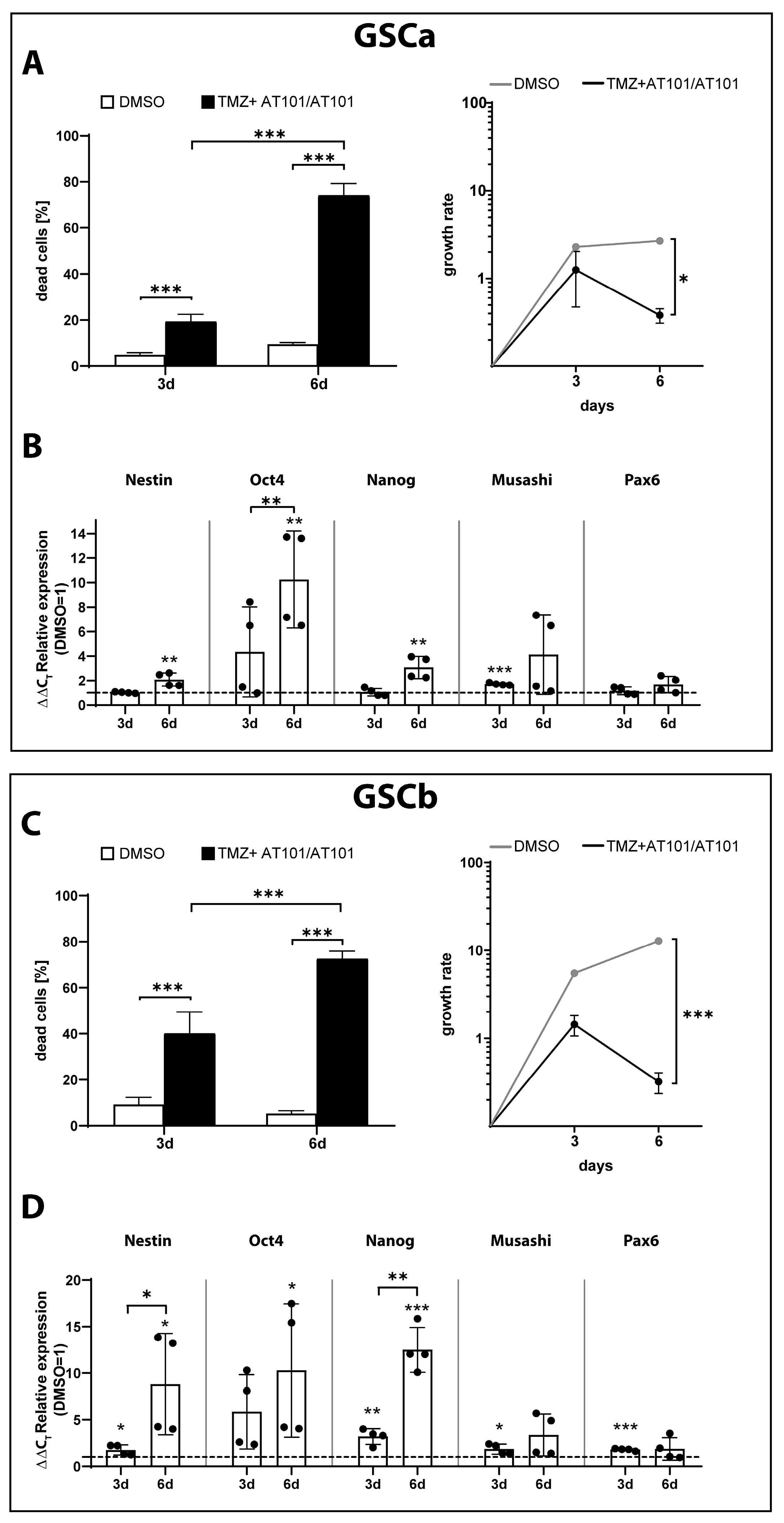
2.3. TMZ+AT101/AT101 Treatment of GSCs
2.4. Torin2-Mediated mTOR Inhibition Counteracts TMZ+AT101/AT101-Regulated Chemoresistance Mechanisms of Surviving GBM Cells
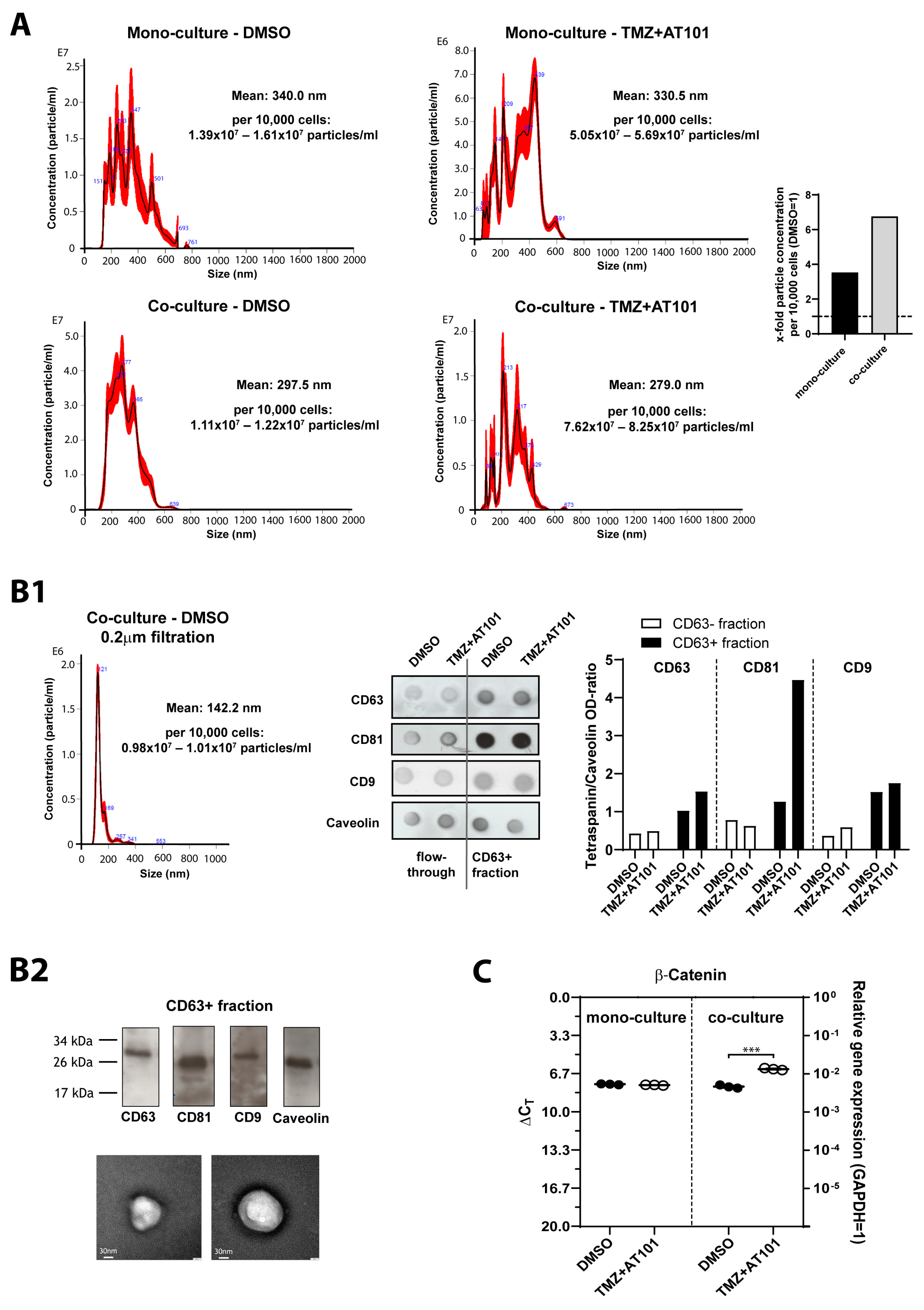
2.5. TMZ+AT101/AT101 Treatment-Influenced Extracellular Vesicles Derived from Surviving GBM Cells
3. Discussion
4. Materials and Methods
4.1. Cultivation of Primary GBM Cells, Patient-Derived Glioma Stem-like Cells (GSCs), and Healthy Brain Cell Lines
4.2. Stimulation of Cells
4.3. Cytotoxicity Assay
4.4. pSIVA Real-Time Apoptosis Microscopy
4.5. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.6. Mitogen-Activated Protein-Kinase (MAPK) Antibody Array
4.7. Western Blot
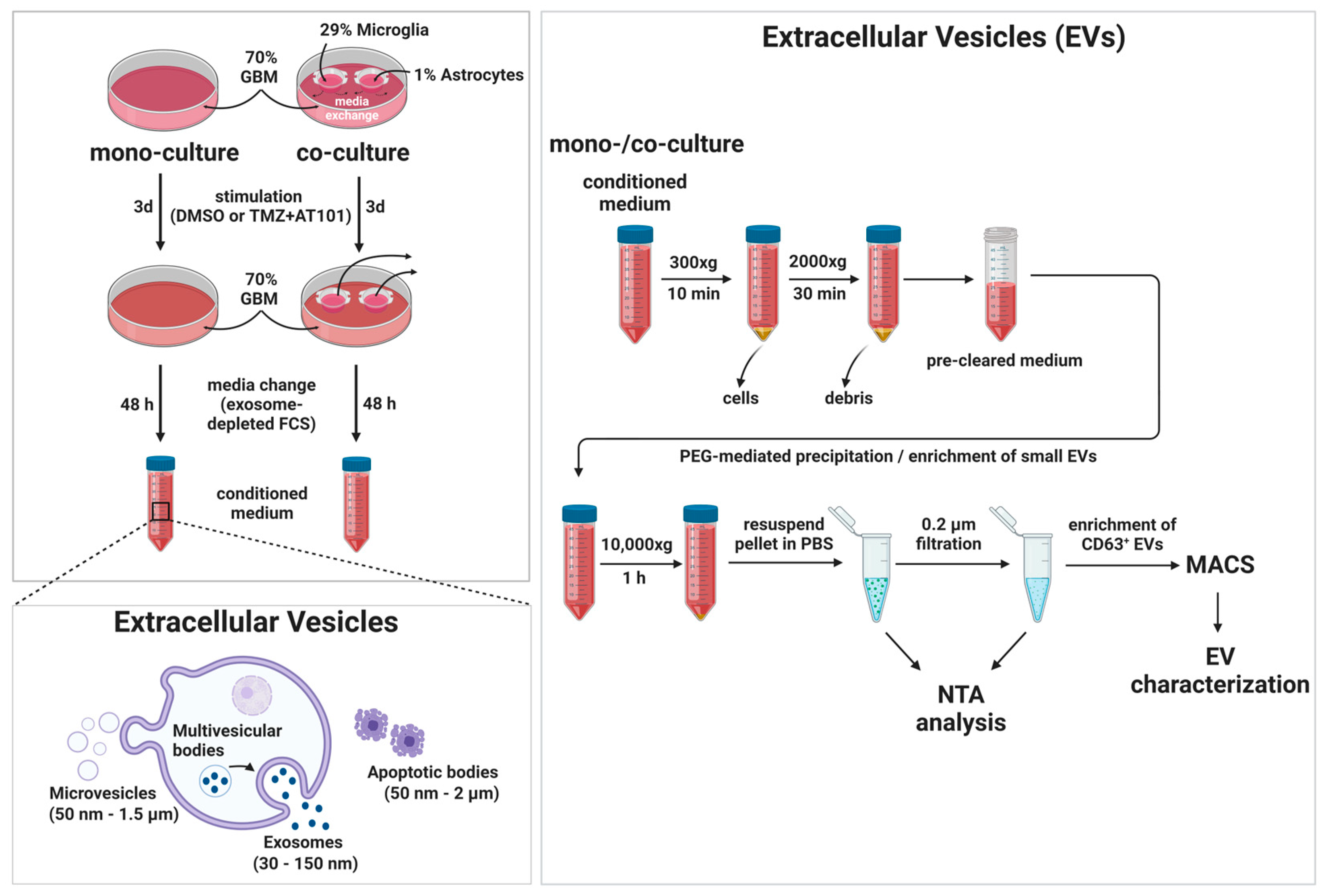
4.8. Separation and Characterization of Extracellular Vesicles (EVs)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, J.; Lv, X.; Lu, C.; Ye, X.; Chen, X.; Fu, J.; Luo, C.-H.; Zhao, Y. Prognostic factors of patients with Gliomas—An analysis on 335 patients with Glioblastoma and other forms of Gliomas. BMC Cancer 2020, 20, 35. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. NeuroMolecular Med. 2018, 20, 437–451. [Google Scholar] [CrossRef]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2021, 7, 620677. [Google Scholar] [CrossRef]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J.; et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 2008, 6816, 6643–6651. [Google Scholar] [CrossRef]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 1296, 829–848. [Google Scholar] [CrossRef]
- Cruz, J.V.R.; Batista, C.; Afonso, B.d.H.; Alexandre-Moreira, M.S.; Dubois, L.G.; Pontes, B.; Neto, V.M.; Mendes, F.D.A. Obstacles to Glioblastoma Treatment Two Decades after Temozolomide. Cancers 2022, 14, 3203. [Google Scholar] [CrossRef]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef]
- Khayamzadeh, M.; Niazi, V.; Hussen, B.M.; Taheri, M.; Ghafouri-Fard, S.; Samadian, M. Emerging role of extracellular vesicles in the pathogenesis of glioblastoma. Metab. Brain Dis. 2022, 38, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Mohtashami, E.; Shafaei-Bajestani, N.; Mollazadeh, H.; Mousavi, S.H.; Jalili-Nik, M.; Sahebkar, A.; Afshari, A.R. The Current State of Potential Therapeutic Modalities for Glioblastoma Multiforme: A Clinical Review. Curr. Drug Metab. 2020, 21, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Asija, S.; Chatterjee, A.; Yadav, S.; Chekuri, G.; Karulkar, A.; Jaiswal, A.K.; Goda, J.S.; Purwar, R. Combinatorial approaches to effective therapy in glioblastoma (GBM): Current status and what the future holds. Int. Rev. Immunol. 2022, 41, 582–605. [Google Scholar] [CrossRef]
- Adamski, V.; Hempelmann, A.; Flüh, C.; Lucius, R.; Synowitz, M.; Hattermann, K.; Held-Feindt, J. Dormant glioblastoma cells acquire stem cell characteristics and are differentially affected by Temozolomide and AT101 treatment. Oncotarget 2017, 8, 108064–108078. [Google Scholar] [CrossRef]
- Adamski, V.; Schmitt, C.; Ceynowa, F.; Adelung, R.; Lucius, R.; Synowitz, M.; Hattermann, K.; Held-Feindt, J. Effects of sequentially applied single and combined temozolomide, hydroxychloroquine and AT101 treatment in a long-term stimulation glioblastoma in vitro model. J. Cancer Res. Clin. Oncol. 2018, 144, 1475–1485. [Google Scholar] [CrossRef]
- Schmitt, C.; Adamski, V.; Rasch, F.; Adelung, R.; Lucius, R.; Synowitz, M.; Hattermann, K.; Held-Feindt, J. Establishment of a glioblastoma in vitro (in)complete resection dual co-culture model suitable for drug testing. Ann. Anat.-Anat. Anz. 2020, 228, 151440. [Google Scholar] [CrossRef]
- Lu, Y.; Li, J.; Dong, C.-E.; Huang, J.; Zhou, H.; Wang, W. Recent advances in gossypol derivatives and analogs: A chemistry and biology view. Futur. Med. Chem. 2017, 9, 1243–1275. [Google Scholar] [CrossRef]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharmacol. 2017, 136, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kögel, D. The Pan-Bcl-2 Inhibitor (−)-Gossypol Triggers Autophagic Cell Death in Malignant Glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Warnsmann, V.; Meyer, N.; Hamann, A.; Kögel, D.; Osiewacz, H.D. A novel role of the mitochondrial permeability transition pore in (−)-gossypol-induced mitochondrial dysfunction. Mech. Ageing Dev. 2018, 170, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Navé, B.T.; Ouwens, M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 1999, 344, 427–431. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef]
- Friedman, M.D.; Jeevan, D.S.; Tobias, M.; Murali, R.; Jhanwar-Uniyal, M. Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid. Oncol. Rep. 2013, 30, 1645–1650. [Google Scholar] [CrossRef]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef]
- Korur, S.; Huber, R.M.; Sivasankaran, B.; Petrich, M.; Morin, P., Jr.; Hemmings, B.A.; Merlo, A.; Lino, M.M. GSK3β Regulates Differentiation and Growth Arrest in Glioblastoma. PLoS ONE 2009, 4, e7443. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.-K.; Ahn, S.H.; Lee, J.; Nam, D.-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2016, 96, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.G.; Jeong, S.W.; Gillick, J.L.; Sursal, T.; Murali, R.; Gandhi, C.D.; Jhanwar-Uniyal, M. Targeting the mTOR pathway using novel ATP-competitive inhibitors, Torin1, Torin2 and XL388, in the treatment of glioblastoma. Int. J. Oncol. 2021, 59, 83. [Google Scholar] [CrossRef] [PubMed]
- Yekula, A.; Taylor, A.; Beecroft, A.; Kang, K.M.; Small, J.L.; Muralidharan, K.; Rosh, Z.; Carter, B.S.; Balaj, L. The role of extracellular vesicles in acquisition of resistance to therapy in glioblastomas. Cancer Drug Resist. 2021, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Kaynak, A.; Davis, H.W.; Kogan, A.B.; Lee, J.-H.; Narmoneva, D.A.; Qi, X. Phosphatidylserine: The Unique Dual-Role Biomarker for Cancer Imaging and Therapy. Cancers 2022, 14, 2536. [Google Scholar] [CrossRef]
- Lee, S.-H.; Meng, X.W.; Flatten, K.S.; Loegering, D.A.; Kaufmann, S.H. Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death Differ. 2013, 20, 64–76. [Google Scholar] [CrossRef]
- Zhao, D.; Stafford, J.H.; Zhou, H.; Thorpe, P.E. Near-infrared Optical Imaging of Exposed Phosphatidylserine in a Mouse Glioma Model. Transl. Oncol. 2011, 4, 355–364. [Google Scholar] [CrossRef]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 2638–2645. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. BioMed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, C.; Kirubakaran, S.; Zhang, X.; Hur, W.; Liu, Y.; Kwiatkowski, N.P.; Wang, J.; Westover, K.D.; Gao, P.; et al. Characterization of Torin2, an ATP-Competitive Inhibitor of mTOR, ATM, and ATRPIKK Inhibitor Torin2 with Antitumor Activity. Cancer Res. 2013, 73, 2574–2586. [Google Scholar] [CrossRef]
- Atkins, R.; Stylli, S.; Luwor, R.; Kaye, A.; Hovens, C. Glycogen synthase kinase-3β (GSK-3β) and its dysregulation in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Chiarini, F.; Paganelli, F.; Marmiroli, S.; Martelli, A.M. Crosstalks of GSK3 signaling with the mTOR network and effects on targeted therapy of cancer. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118635. [Google Scholar] [CrossRef] [PubMed]
- Kubelt, C.; Hattermann, K.; Sebens, S.; Mehdorn, H.M.; Held-Feindt, J. Epithelial-to-mesenchymal transition in paired human primary and recurrent glioblastomas. Int. J. Oncol. 2015, 46, 2515–2525. [Google Scholar] [CrossRef]
- Hattermann, K.; Flüh, C.; Engel, D.; Mehdorn, H.M.; Synowitz, M.; Mentlein, R.; Held-Feindt, J. Stem cell markers in glioma progression and recurrence. Int. J. Oncol. 2016, 49, 1899–1910. [Google Scholar] [CrossRef]
- Joshi, K.; Banasavadi-Siddegowda, Y.; Mo, X.; Kim, S.-H.; Mao, P.; Kig, C.; Nardini, D.; Sobol, R.W.; Chow, L.M.; Kornblum, H.I.; et al. MELK-Dependent FOXM1 Phosphorylation is Essential for Proliferation of Glioma Stem Cells. Stem Cells 2013, 31, 1051–1063. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, Z.; Shen, L.; Tang, T.; Yang, M.; Zheng, X. Long Noncoding RNA LINC-PINT Suppresses Cell Proliferation, Invasion, and EMT by Blocking Wnt/β-Catenin Signaling in Glioblastoma. Front. Pharmacol. 2021, 11, 11. [Google Scholar] [CrossRef]
- Chen, W.; Kong, K.-K.; Xu, X.-K.; Chen, C.; Li, H.; Wang, F.-Y.; Peng, X.-F.; Zhang, Z.; Li, P.; Li, J.-L.; et al. Downregulation of miR-205 is associated with glioblastoma cell migration, invasion, and the epithelial-mesenchymal transition, by targeting ZEB1 via the Akt/mTOR signaling pathway. Int. J. Oncol. 2018, 52, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Zheng, Y.; Zhao, Y.; Xiu, X.; Wang, J. miR-590-3p suppresses cancer cell migration, invasion and epithelial–mesenchymal transition in glioblastoma multiforme by targeting ZEB1 and ZEB2. Biochem. Biophys. Res. Commun. 2015, 468, 739–745. [Google Scholar] [CrossRef]
- Mentlein, R.; Forstreuter, F.; Mehdorn, H.M.; Held-Feindt, J. Functional Significance of Vascular Endothelial Growth Factor Receptor Expression on Human Glioma Cells. J. Neuro-Oncol. 2004, 67, 9–18. [Google Scholar] [CrossRef]
- Gousias, K.; Theocharous, T.; Simon, M. Mechanisms of Cell Cycle Arrest and Apoptosis in Glioblastoma. Biomedicines 2022, 10, 564. [Google Scholar] [CrossRef]
- Huang, M.; Ke, Y.; Sun, X.; Yu, L.; Yang, Z.; Zhang, Y.; DU, M.; Wang, J.; Liu, X.; Huang, S. Mammalian target of rapamycin signaling is involved in the vasculogenic mimicry of glioma via hypoxia-inducible factor-1α. Oncol. Rep. 2014, 32, 1973–1980. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Pandolfi, P.P. The Multiple Roles of PTEN in Tumor Suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef]
- Flügel, D.; Görlach, A.; Michiels, C.; Kietzmann, T. Glycogen Synthase Kinase 3 Phosphorylates Hypoxia-Inducible Factor 1α and Mediates Its Destabilization in a VHL-Independent Manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- de Andrade, K.C.; Lee, E.E.; Tookmanian, E.M.; Kesserwan, C.A.; Manfredi, J.J.; Hatton, J.N.; Loukissas, J.K.; Zavadil, J.; Zhou, L.; Olivier, M.; et al. The TP53 Database: Transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022, 29, 1071–1073. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Beuvink, I.; Boulay, A.; Fumagalli, S.; Zilbermann, F.; Ruetz, S.; O’Reilly, T.; Natt, F.; Hall, J.; Lane, H.A.; Thomas, G. The mTOR Inhibitor RAD001 Sensitizes Tumor Cells to DNA-Damaged Induced Apoptosis through Inhibition of p21 Translation. Cell 2005, 120, 747–759. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Llanos, S.; García-Pedrero, J.M.; Morgado-Palacin, L.; Rodrigo, J.P.; Serrano, M. Stabilization of p21 by mTORC1/4E-BP1 predicts clinical outcome of head and neck cancers. Nat. Commun. 2016, 7, 10438. [Google Scholar] [CrossRef] [PubMed]
- Tahmasebi, S.; Alain, T.; Rajasekhar, V.K.; Zhang, J.-P.; Prager-Khoutorsky, M.; Khoutorsky, A.; Dogan, Y.; Gkogkas, C.G.; Petroulakis, E.; Sylvestre, A.; et al. Multifaceted Regulation of Somatic Cell Reprogramming by mRNA Translational Control. Cell Stem Cell 2014, 14, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Caylioglu, D.; Meyer, R.J.; Hellmold, D.; Kubelt, C.; Synowitz, M.; Held-Feindt, J. Effects of the Anti-Tumorigenic Agent AT101 on Human Glioblastoma Cells in the Microenvironmental Glioma Stem Cell Niche. Int. J. Mol. Sci. 2021, 22, 3606. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Blandino, G.; Deppert, W.; Hainaut, P.; Levine, A.; Lozano, G.; Olivier, M.; Rotter, V.; Wiman, K.; Oren, M. Mutant p53 protein, master regulator of human malignancies: A report on the fifth Mutant p53 Workshop. Cell Death Differ. 2012, 19, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Pongor, L.; Su, Y.-T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res./Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, F.; Zhou, Y.; Chen, W.; Shao, H.; Zhang, Y. MGMT Promoter Methylation and Glioblastoma Prognosis: A Systematic Review and Meta-analysis. Arch. Med. Res. 2013, 44, 281–290. [Google Scholar] [CrossRef]
- Wick, W.; Platten, M. Understanding and Targeting Alkylator Resistance in Glioblastoma. Cancer Discov. 2014, 4, 1120–1122. [Google Scholar] [CrossRef]
- Alexander, B.M.; Ligon, K.L.; Wen, P.Y. Enhancing radiation therapy for patients with glioblastoma. Expert Rev. Anticancer. Ther. 2013, 13, 569–581. [Google Scholar] [CrossRef]
- Mc Namee, N.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Panzarini, E.; Tacconi, S.; Carata, E.; Mariano, S.; Tata, A.M.; Dini, L. Molecular Characterization of Temozolomide-Treated and Non Temozolomide-Treated Glioblastoma Cells Released Extracellular Vesicles and Their Role in the Macrophage Response. Int. J. Mol. Sci. 2020, 21, 8353. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Ariotti, N.; Wu, Y.; Okano, S.; Gambin, Y.; Follett, J.; Rae, J.; Ferguson, C.; Teasdale, R.D.; Alexandrov, K.; Meunier, F.A.; et al. An inverted CAV1 (caveolin 1) topology defines novel autophagy-dependent exosome secretion from prostate cancer cells. Autophagy 2021, 17, 2200–2216. [Google Scholar] [CrossRef]
- Campos, A.; Salomon, C.; Bustos, R.; Díaz, J.; Martínez, S.; Silva, V.; Reyes, C.; Díiaz-Valdivia, N.; Varas-Godoy, M.; Lobos-González, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine 2018, 13, 2597–2609. [Google Scholar] [CrossRef]
- Kıyga, E.; Adıgüzel, Z.; Uçar, E. Temozolomide increases heat shock proteins in extracellular vesicles released from glioblastoma cells. Mol. Biol. Rep. 2022, 49, 8701–8713. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Maire, C.L.; Reimer, R.; Dührsen, L.; Kolbe, K.; Holz, M.; Schneider, E.; Rissiek, A.; Babayan, A.; Hille, C.; et al. Imaging flow cytometry facilitates multiparametric characterization of extracellular vesicles in malignant brain tumours. J. Extracell. Vesicles 2019, 8, 1588555. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 1138, E968–E977. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Stevic, I.; Buescher, G.; Ricklefs, F.L. Monitoring Therapy Efficiency in Cancer through Extracellular Vesicles. Cells 2020, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Maire, C.L.; Matschke, J.; Dührsen, L.; Sauvigny, T.; Holz, M.; Kolbe, K.; Peine, S.; Herold-Mende, C.; Carter, B.; et al. FASN Is a Biomarker Enriched in Malignant Glioma-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 1931. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- André-Grégoire, G.; Bidère, N.; Gavard, J. Temozolomide affects Extracellular Vesicles Released by Glioblastoma Cells. Biochimie 2018, 155, 11–15. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Ayroldi, E.; Giusti, I.; Dolo, V.; Cifone, M.G.; Cinque, B.; Palumbo, P. Cyclooxygenase-2 Upregulated by Temozolomide in Glioblastoma Cells Is Shuttled In Extracellular Vesicles Modifying Recipient Cell Phenotype. Front. Oncol. 2022, 12, 933746. [Google Scholar] [CrossRef]
- Ma, Y.; Yoshida, T.; Matoba, K.; Kida, K.; Shintani, R.; Piao, Y.; Jin, J.; Nishino, T.; Hanayama, R. Identification of small compounds regulating the secretion of extracellular vesicles via a TIM4-affinity ELISA. Sci. Rep. 2021, 11, 13471. [Google Scholar] [CrossRef]
- Pan, J.; Sheng, S.; Ye, L.; Xu, X.; Ma, Y.; Feng, X.; Qiu, L.; Fan, Z.; Wang, Y.; Xia, X.; et al. Extracellular vesicles derived from glioblastoma promote proliferation and migration of neural progenitor cells via PI3K-Akt pathway. Cell Commun. Signal. 2022, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Flüh, C.; Chitadze, G.; Adamski, V.; Hattermann, K.; Synowitz, M.; Kabelitz, D.; Held-Feindt, J. NKG2D ligands in glioma stem-like cells: Expression in situ and in vitro. Histochem. Cell Biol. 2018, 149, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Hattermann, K.; Held-Feindt, J.; Lucius, R.; Müerköster, S.S.; Penfold, M.E.; Schall, T.J.; Mentlein, R. The Chemokine Receptor CXCR7 Is Highly Expressed in Human Glioma Cells and Mediates Antiapoptotic Effects. Cancer Res. 2010, 70, 3299–3308. [Google Scholar] [CrossRef] [PubMed]
- Held-Feindt, J.; Schmelz, S.; Hattermann, K.; Mentlein, R.; Mehdorn, H.M.; Sebens, S. The neural adhesion molecule L1CAM confers chemoresistance in human glioblastomas. Neurochem. Int. 2012, 61, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.; Tylden, G.D.; Dumoulin, A.; Sharma, B.N.; Hirsch, H.H.; Rinaldo, C.H. The Human Fetal Glial Cell Line SVG p12 Contains Infectious BK Polyomavirus. J. Virol. 2014, 88, 7556–7568. [Google Scholar] [CrossRef]
- Schweighardt, J.T.S.B.; Shieh, J.T.C.; Atwood, W.J. CD4/CXCR4-independent infection of human astrocytes by a T-tropic strain of HIV-1. J. Neurovirology 2001, 7, 155–162. [Google Scholar] [CrossRef]
- Adamski, V.; Hattermann, K.; Kubelt, C.; Cohrs, G.; Lucius, R.; Synowitz, M.; Sebens, S.; Held-Feindt, J. Entry and exit of chemotherapeutically-promoted cellular dormancy in glioblastoma cells is differentially affected by the chemokines CXCL12, CXCL16, and CX3CL1. Oncogene 2020, 22, 4421–4435. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hellmold, D.; Kubelt, C.; Daunke, T.; Beckinger, S.; Janssen, O.; Hauck, M.; Schütt, F.; Adelung, R.; Lucius, R.; Haag, J.; et al. Sequential Treatment with Temozolomide Plus Naturally Derived AT101 as an Alternative Therapeutic Strategy: Insights into Chemoresistance Mechanisms of Surviving Glioblastoma Cells. Int. J. Mol. Sci. 2023, 24, 9075. https://doi.org/10.3390/ijms24109075
Hellmold D, Kubelt C, Daunke T, Beckinger S, Janssen O, Hauck M, Schütt F, Adelung R, Lucius R, Haag J, et al. Sequential Treatment with Temozolomide Plus Naturally Derived AT101 as an Alternative Therapeutic Strategy: Insights into Chemoresistance Mechanisms of Surviving Glioblastoma Cells. International Journal of Molecular Sciences. 2023; 24(10):9075. https://doi.org/10.3390/ijms24109075
Chicago/Turabian StyleHellmold, Dana, Carolin Kubelt, Tina Daunke, Silje Beckinger, Ottmar Janssen, Margarethe Hauck, Fabian Schütt, Rainer Adelung, Ralph Lucius, Jochen Haag, and et al. 2023. "Sequential Treatment with Temozolomide Plus Naturally Derived AT101 as an Alternative Therapeutic Strategy: Insights into Chemoresistance Mechanisms of Surviving Glioblastoma Cells" International Journal of Molecular Sciences 24, no. 10: 9075. https://doi.org/10.3390/ijms24109075
APA StyleHellmold, D., Kubelt, C., Daunke, T., Beckinger, S., Janssen, O., Hauck, M., Schütt, F., Adelung, R., Lucius, R., Haag, J., Sebens, S., Synowitz, M., & Held-Feindt, J. (2023). Sequential Treatment with Temozolomide Plus Naturally Derived AT101 as an Alternative Therapeutic Strategy: Insights into Chemoresistance Mechanisms of Surviving Glioblastoma Cells. International Journal of Molecular Sciences, 24(10), 9075. https://doi.org/10.3390/ijms24109075