
Protein Posttranslational Modification in Stemness Remodeling and Its Emerging Role as a Novel Therapeutic Target in Gastrointestinal Cancers

Abstract
:1. Introduction
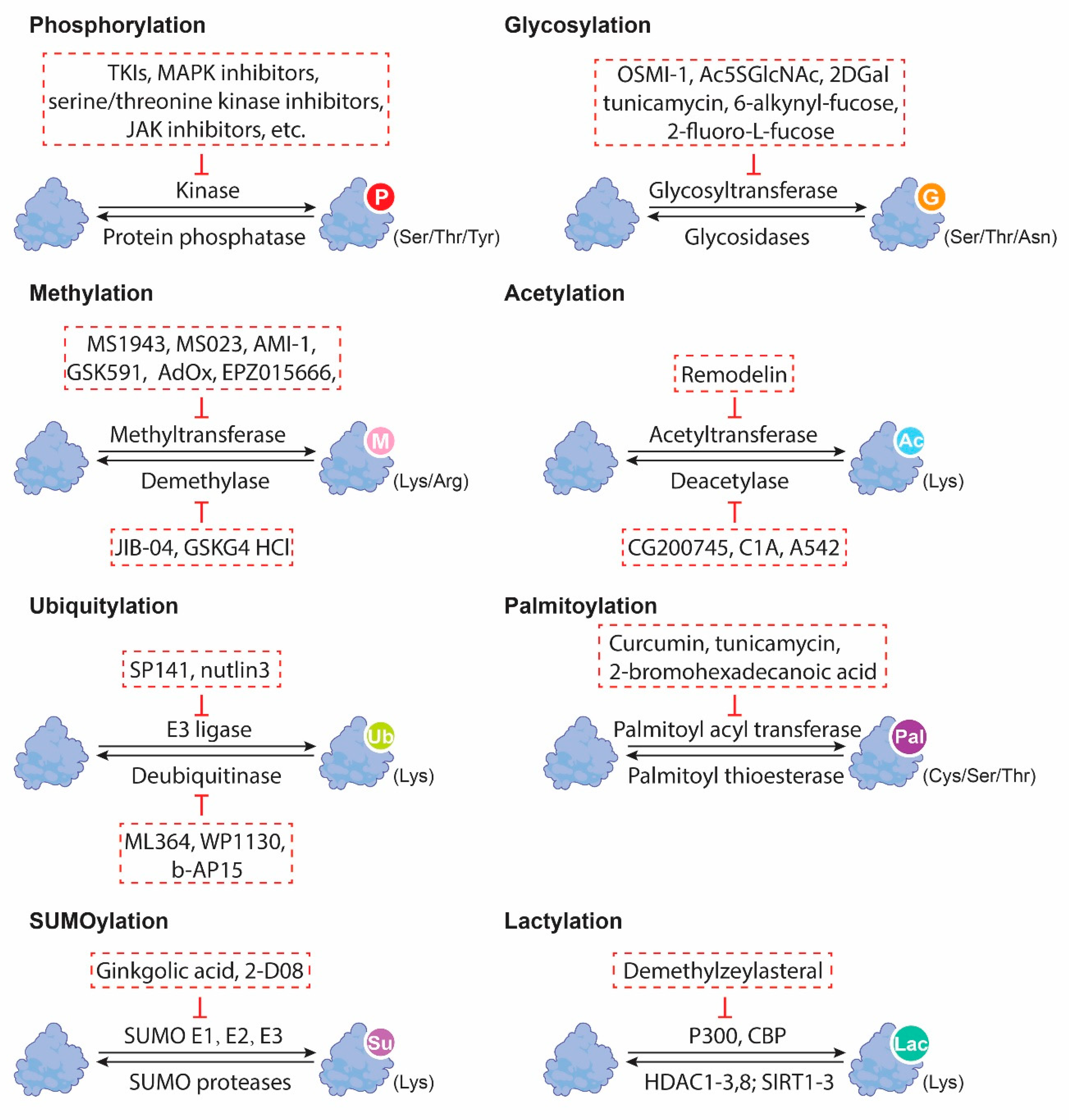
2. Overview of Protein PTMs
3. Cancer Stemness
4. PTMs and Cancer Stemness in GI Malignancies
4.1. Protein Phosphorylation and Cancer Stemness
4.2. Protein Glycosylation and Cancer Stemness
4.3. Protein Methylation and Cancer Stemness
4.4. Protein Acetylation and Cancer Stemness
4.5. Others PTMs and Cancer Stemness
5. The Crosstalk of Intracellular Protein PTMs on Stemness
6. Therapeutic Targeting of PTMs in GI CSCs

{kind=link}
{kind=link}
| Cancer Type | PTM Types | Proteins Targets | Mechanisms | Ref |
|---|---|---|---|---|
| EC | Phosphorylation | STAT3 | Phosphorylation of STAT3 induces NANOG upregulation. | [16] |
| p53 | Nuclear phosphorylated p53 bound to NANOG promoter to inhibit OCT4- and SOX2-induced NANOG transcription. | [34] | ||
| Glycosylation | -- | OGT depletion attenuated the self-renewal and tumorigenic capacities of ALDH+ esophageal CSCs. | [42] | |
| HCC | Deacetylation | β-catenin | Deacetylation of β-catenin caused by BBOX1 degradation transactivated SOX2, OCT4, and NANOG. | [61] |
| Glycosylation | -- | Knockdown of glycosylation-associated genes (e.g., CAD, SLC51B, LGALS3, B3GAT3, and MT3) led to decreased stemness markers CD24, CD44, CD20, FOXM1, and EpCAM. | [37] | |
| CD147, ICAM-1, EGFR, EPHA2 | Under glucose restriction, FUT1-induced α1,2-fucosylation on CD147, ICAM-1, EGFR, and EPHA2 dysregulated AKT/mTOR/4EBP1 signaling to potentiate stemness. | [10] | ||
| eIF4E | O-GlcNAcylatd eIF4E directly interacted with 5′UTR of SOX2 to result in an enrichment of CD133+ hepatoma cells. | [41] | ||
| Methylation | CRAF | PRMT6 methylated CRAF on arginine 100 to suppress the RAS/RAF binding and MEK/ERK axis. | [44] | |
| Palmitoylation | α-enolase, c-Myc promoter-binding protein 1, etc. | Palmitate stimulated the palmitoylation of these proteins for enhanced sphere-forming capability. | [59] | |
| SUMOylation | HIF-1α | SUMO protease SENP1 and HIF-1α formed a positive feedback loop to expand CD24+ subpopulation. | [57] | |
| PC | Glycosylation | -- | ST6Gal1 induced stem cell transcription factors Sox9 and Slug. | [40] |
| -- | Knockout of OGT inhibited the tumor-initiating capacity. | [8] | ||
| SOX2 | O-GlcNAcylation of SOX2 at S246A by OGT increased its stability in the nucleus. | [42] | ||
| CRC | Phosphorylation | YAP | Dephosphorylated YAP contributed to stiff-matrix-potentiated CRC stemness features. | [18] |
| Methylation | YAP | Methylation of YAP at K342 induced c-Met upregulation for stemness features. | [46] | |
| Ubiquitination | p53 | p53 ubiquitination caused p53 destabilization and further stemness features. | [56]. | |
| Glycosylation | -- | FUT9-mediated hyperfucosylation also triggered Sox2, ALDH, and CD44 expression and tumor sphere formation. | [9] | |
| Fas receptors | ST6Gal1 loss reduced the CD133+/ALDH1+ CSCs population. | [7] | ||
| -- | Global O-GlcNAcylation downregulation led to increased CD133 and CD44 expression and enhanced spheroid-forming ability. | [43] | ||
| Lactylation | β-catenin | β-catenin lactylation stimulated the Wnt signaling pathway to potentiate CRC stemness. | [58] | |
| GC | Phosphorylation | YAP | SCD1 induced YAP phosphorylation to affect cell stemness. | [29] |
| Cancer Type | PTM Types | Proteins Targets | Mechanisms | Ref |
|---|---|---|---|---|
| EC | Methylation | NOTCH1 | Demethylated NOTCH1 at its promoters H3K9me2 and H3K9me3. | [49] |
| histone H4 | Demethylation of histone H4 arginine catalyzed by PRMT1 activated Wnt/β-catenin and Notch signaling pathways. | [45] | ||
| H3K9me2, H3K9me3 | GASC1 methylated H3K9me2 and H3K9me3 on NOTCH1 promoter to reduce NOTCH1, leading to impaired stemness. | [49] | ||
| HCC | Methylation | Prickle1, APC | Methylation of Prickle1 and APC promoters activated β-catenin signaling and subsequent stem-like characteristics. | [11] |
| Deacetylation | LKB1 | HDAC11 mediated histone deacetylation at the promoter region of LKB1 to inhibit LKB1 transcription, which further abrogated AMPK signaling and accelerated glycolysis to maintain CSCs. | [52] | |
| CRC | Methylation | H3K79 | H3K79 methylation contributed to IL-22-induced transcriptional upregulation of STAT3 and subsequent stemness features. | [47] |
| H3K4me3, H3K27me3 | Upregulated histone H3K4me3 and downregulated histone H3K27me3 collaboratively induced transactivation of CD133, CD44, and ALDH1A1 by binding to their promoters. | [48] | ||
| Acetylation | H3 lysine 9 | SIRT1 deacetylated H3 lysine 9 on miR-1185-1 promoter to repress its expression and disrupted its targeting on CD24 3′UTR. | [51] |
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ullah, I.; Yang, L.; Yin, F.-T.; Sun, Y.; Li, X.-H.; Li, J.; Wang, X. Multi-Omics Approaches in Colorectal Cancer Screening and Diagnosis, Recent Updates and Future Perspectives. Cancers 2022, 14, 5545. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Croonenborghs, T.; Roca Suarez, A.A.; Van Renne, N.; Juhling, F.; Oudot, M.A.; Virzi, A.; Bandiera, S.; Jamey, C.; Meszaros, G.; et al. Combined Analysis of Metabolomes, Proteomes, and Transcriptomes of Hepatitis C Virus-Infected Cells and Liver to Identify Pathways Associated With Disease Development. Gastroenterology 2019, 157, 537–551.e539. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Barkeer, S.; Chugh, S.; Batra, S.K.; Ponnusamy, M.P. Glycosylation of Cancer Stem Cells: Function in Stemness, Tumorigenesis, and Metastasis. Neoplasia 2018, 20, 813–825. [Google Scholar] [CrossRef]
- Swindall, A.F.; Londono-Joshi, A.I.; Schultz, M.J.; Fineberg, N.; Buchsbaum, D.J.; Bellis, S.L. ST6Gal-I protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res. 2013, 73, 2368–2378. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, L.; Ge, D.; Tan, L.; Cao, B.; Fan, H.; Xue, L. Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8+ T cells. Cancer Lett. 2021, 500, 98–106. [Google Scholar] [CrossRef]
- Blanas, A.; Zaal, A.; van der Haar Avila, I.; Kempers, M.; Kruijssen, L.; de Kok, M.; Popovic, M.A.; van der Horst, J.; van Vliet, S.J. FUT9-Driven Programming of Colon Cancer Cells towards a Stem Cell-Like State. Cancers 2020, 12, 2580. [Google Scholar] [CrossRef]
- Loong, J.H.; Wong, T.L.; Tong, M.; Sharma, R.; Zhou, L.; Ng, K.Y.; Yu, H.J.; Li, C.H.; Man, K.; Lo, C.M.; et al. Glucose deprivation-induced aberrant FUT1-mediated fucosylation drives cancer stemness in hepatocellular carcinoma. J. Clin. Investig. 2021, 131, e143377. [Google Scholar] [CrossRef]
- Lei, Z.J.; Wang, J.; Xiao, H.L.; Guo, Y.; Wang, T.; Li, Q.; Liu, L.; Luo, X.; Fan, L.L.; Lin, L.; et al. Lysine-specific demethylase 1 promotes the stemness and chemoresistance of Lgr5+ liver cancer initiating cells by suppressing negative regulators of beta-catenin signaling. Oncogene 2015, 34, 3188–3198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhuang, H.; Cao, F.; Li, J.; Guo, Y.; Zhang, J.; Zhao, Q.; Liu, Y. Shc3 promotes hepatocellular carcinoma stemness and drug resistance by interacting with beta-catenin to inhibit its ubiquitin degradation pathway. Cell Death Dis. 2021, 12, 278. [Google Scholar] [CrossRef]
- Zhou, S.; Peng, J.; Xiao, L.; Zhou, C.; Fang, Y.; Ou, Q.; Qin, J.; Liu, M.; Pan, Z.; Hou, Z. TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell Death Dis. 2021, 12, 463. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef]
- Regnier, F.E.; Kim, J. Proteins and Proteoforms: New Separation Challenges. Anal. Chem. 2018, 90, 361–373. [Google Scholar] [CrossRef]
- Nishi, H.; Hashimoto, K.; Panchenko, A.R. Phosphorylation in protein-protein binding: Effect on stability and function. Structure 2011, 19, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Mann, M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 2010, 11, 427–439. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, F.; Zhang, X.; Lin, H.K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target. Ther. 2021, 6, 422. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.L.; Che, N.; Ma, S. Reprogramming of central carbon metabolism in cancer stem cells. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Lathia, J.D.; Liu, H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target. Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Krueger, K.E.; Srivastava, S. Posttranslational protein modifications: Current implications for cancer detection, prevention, and therapeutics. Mol. Cell. Proteom. 2006, 5, 1799–1810. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Xi, H.; Cui, J.; Zhang, K.; Zhang, J.; Zhang, Y.; Xu, W.; Liang, W.; Zhuang, Z.; et al. Stearoyl-CoA-desaturase-1 regulates gastric cancer stem-like properties and promotes tumour metastasis via Hippo/YAP pathway. Br. J. Cancer 2020, 122, 1837–1847. [Google Scholar] [CrossRef]
- Yu, H.; Zhou, L.; Loong, J.H.C.; Lam, K.H.; Wong, T.L.; Ng, K.Y.; Tong, M.; Ma, V.W.; Wang, Y.; Zhang, X.; et al. SERPINA12 promotes the tumorigenic capacity of HCC stem cells through hyperactivation of AKT/beta-catenin signaling. Hepatology 2023. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; Graveel, C.R.; Zylstra-Diegel, C.R.; Zhong, Z.; Williams, B.O. Wnt/beta-catenin Signaling in Normal and Cancer Stem Cells. Cancers 2011, 3, 2050–2079. [Google Scholar] [CrossRef]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Huang, Y.; Pei, Q.; Liu, H.; Pei, H.; Zhu, H. Matrix stiffness mediates stemness characteristics via activating the Yes-associated protein in colorectal cancer cells. J. Cell. Biochem. 2019, 120, 2213–2225. [Google Scholar] [CrossRef]
- Zhao, Q.W.; Zhou, Y.W.; Li, W.X.; Kang, B.; Zhang, X.Q.; Yang, Y.; Cheng, J.; Yin, S.Y.; Tong, Y.; He, J.Q.; et al. Akt-mediated phosphorylation of Oct4 is associated with the proliferation of stem-like cancer cells. Oncol. Rep. 2015, 33, 1621–1629. [Google Scholar] [CrossRef]
- Liu, P.; Zhou, Q.; Li, J. Integrated Multi-Omics Data Analysis Reveals Associations Between Glycosylation and Stemness in Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 913432. [Google Scholar] [CrossRef]
- Moriwaki, K.; Okudo, K.; Haraguchi, N.; Takeishi, S.; Sawaki, H.; Narimatsu, H.; Tanemura, M.; Ishii, H.; Mori, M.; Miyoshi, E. Combination use of anti-CD133 antibody and SSA lectin can effectively enrich cells with high tumorigenicity. Cancer Sci. 2011, 102, 1164–1170. [Google Scholar] [CrossRef]
- Azuma, K.; Serada, S.; Takamatsu, S.; Terao, N.; Takeishi, S.; Kamada, Y.; Naka, T.; Miyoshi, E. Identification of sialylated glycoproteins in Doxorubicin-treated hepatoma cells with glycoproteomic analyses. J. Proteome Res. 2014, 13, 4869–4877. [Google Scholar] [CrossRef]
- Schultz, M.J.; Holdbrooks, A.T.; Chakraborty, A.; Grizzle, W.E.; Landen, C.N.; Buchsbaum, D.J.; Conner, M.G.; Arend, R.C.; Yoon, K.J.; Klug, C.A.; et al. The Tumor-Associated Glycosyltransferase ST6Gal-I Regulates Stem Cell Transcription Factors and Confers a Cancer Stem Cell Phenotype. Cancer Res. 2016, 76, 3978–3988. [Google Scholar] [CrossRef]
- Cao, B.; Duan, M.; Xing, Y.; Liu, C.; Yang, F.; Li, Y.; Yang, T.; Wei, Y.; Gao, Q.; Jiang, J. O-GlcNAc transferase activates stem-like cell potential in hepatocarcinoma through O-GlcNAcylation of eukaryotic initiation factor 4E. J. Cell. Mol. Med. 2019, 23, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Gupta, V.K.; Dauer, P.; Kesh, K.; Hadad, R.; Giri, B.; Chandra, A.; Dudeja, V.; Slawson, C.; Banerjee, S.; et al. O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability. Theranostics 2019, 9, 3410–3424. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Garcia, G.; Castaneda-Patlan, M.C.; Vercoutter-Edouart, A.S.; Lefebvre, T.; Robles-Flores, M. O-GlcNAcylation Is Involved in the Regulation of Stem Cell Markers Expression in Colon Cancer Cells. Front. Endocrinol. 2019, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.H.; Zhou, L.; Ng, K.Y.; Wong, T.L.; Lee, T.K.; Sharma, R.; Loong, J.H.; Ching, Y.P.; Yuan, Y.F.; Xie, D.; et al. PRMT6 Regulates RAS/RAF Binding and MEK/ERK-Mediated Cancer Stemness Activities in Hepatocellular Carcinoma through CRAF Methylation. Cell Rep. 2018, 25, 690–701.e698. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, Q.; Li, C.; Wang, X.; Jiang, L.; Huang, L.; Wang, C.; Chen, H. PRMT1 regulates the tumour-initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death Dis. 2019, 10, 359. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, Y.; Wei, L.; Wu, S.; Shen, K.; Liu, C.; Dong, Y.; Zhao, Y.; Zhang, Y.; Zhang, C.; et al. ABHD5 inhibits YAP-induced c-Met overexpression and colon cancer cell stemness via suppressing YAP methylation. Nat. Commun. 2021, 12, 6711. [Google Scholar] [CrossRef]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef]
- Ji, G.; Zhou, W.; Du, J.; Zhou, J.; Wu, D.; Zhao, M.; Yang, L.; Hao, A. PCGF1 promotes epigenetic activation of stemness markers and colorectal cancer stem cell enrichment. Cell Death Dis. 2021, 12, 633. [Google Scholar] [CrossRef]
- Jia, R.; Yang, L.; Yuan, X.; Kong, J.; Liu, Y.; Yin, W.; Gao, S.; Zhang, Y. GASC1 Promotes Stemness of Esophageal Squamous Cell Carcinoma via NOTCH1 Promoter Demethylation. J. Oncol. 2019, 2019, 1621054. [Google Scholar] [CrossRef]
- Liu, J.; Qiu, J.; Zhang, Z.; Zhou, L.; Li, Y.; Ding, D.; Zhang, Y.; Zou, D.; Wang, D.; Zhou, Q.; et al. SOX4 maintains the stemness of cancer cells via transcriptionally enhancing HDAC1 revealed by comparative proteomics study. Cell Biosci. 2021, 11, 23. [Google Scholar] [CrossRef]
- Wang, T.W.; Chern, E.; Hsu, C.W.; Tseng, K.C.; Chao, H.M. SIRT1-Mediated Expression of CD24 and Epigenetic Suppression of Novel Tumor Suppressor miR-1185-1 Increases Colorectal Cancer Stemness. Cancer Res. 2020, 80, 5257–5269. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Ren, Y.; Feng, M.; Meng, P.; Wang, Q.; Chen, W.; Jiao, Q.; Wang, Y.; Du, L.; Zhou, F.; et al. HDAC11 Regulates Glycolysis through the LKB1/AMPK Signaling Pathway to Maintain Hepatocellular Carcinoma Stemness. Cancer Res. 2021, 81, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Chen, P.; Yan, H.X.; Fu, G.B.; Luo, F.F.; Zhang, J.; Zhao, S.M.; Zhai, B.; Yu, J.H.; Chen, L.; et al. Targeting mTORC2/HDAC3 Inhibits Stemness of Liver Cancer Cells Against Glutamine Starvation. Adv. Sci. 2022, 9, e2103887. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Zhang, X.; Yao, Q. The F-box protein FBXO11 restrains hepatocellular carcinoma stemness via promotion of ubiquitin-mediated degradation of Snail. FEBS Open Bio 2020, 10, 1810–1820. [Google Scholar] [CrossRef]
- Qin, B.; Zou, S.; Li, K.; Wang, H.; Wei, W.; Zhang, B.; Xiao, L.; Choi, H.H.; Tang, Q.; Huang, D.; et al. CSN6-TRIM21 axis instigates cancer stemness during tumorigenesis. Br. J. Cancer 2020, 122, 1673–1685. [Google Scholar] [CrossRef]
- Yao, J.; Wang, X.P.; Yang, J.; Yang, Z.; Zhang, Z.Y. SCF-FBXL8 contributes to liver metastasis and stem-cell-like features in colorectal cancer cells by mediating ubiquitination and degradation of TP53. Clin. Transl. Med. 2023, 13, e1208. [Google Scholar] [CrossRef]
- Cui, C.P.; Wong, C.C.; Kai, A.K.; Ho, D.W.; Lau, E.Y.; Tsui, Y.M.; Chan, L.K.; Cheung, T.T.; Chok, K.S.; Chan, A.C.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1alpha deSUMOylation and SENP1/HIF-1alpha positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef]
- Miao, Z.; Zhao, X.; Liu, X. Hypoxia induced beta-catenin lactylation promotes the cell proliferation and stemness of colorectal cancer through the wnt signaling pathway. Exp. Cell Res. 2023, 422, 113439. [Google Scholar] [CrossRef]
- Chong, L.W.; Tsai, C.L.; Yang, K.C.; Liao, C.C.; Hsu, Y.C. Targeting protein palmitoylation decreases palmitate-induced sphere formation of human liver cancer cells. Mol. Med. Rep. 2020, 22, 939–947. [Google Scholar] [CrossRef]
- Luk, S.T.; Ng, K.Y.; Zhou, L.; Tong, M.; Wong, T.L.; Yu, H.; Lo, C.M.; Man, K.; Guan, X.Y.; Lee, T.K.; et al. Deficiency in Embryonic Stem Cell Marker Reduced Expression 1 Activates Mitogen-Activated Protein Kinase Kinase 6-Dependent p38 Mitogen-Activated Protein Kinase Signaling to Drive Hepatocarcinogenesis. Hepatology 2020, 72, 183–197. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Zhang, D.; Zhao, W.; Lu, Y.; Liu, C.; Lin, W.; Zhang, Y.; Chen, K.; Wang, H.; et al. CRIP1 suppresses BBOX1-mediated carnitine metabolism to promote stemness in hepatocellular carcinoma. EMBO J. 2022, 41, e110218. [Google Scholar] [CrossRef]
- Zhang, X.; Du, R.; Luo, N.; Xiang, R.; Shen, W. Aspirin mediates histone methylation that inhibits inflammation-related stemness gene expression to diminish cancer stemness via COX-independent manner. Stem Cell Res. Ther. 2020, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Cho, H.I.; Yoon, H.J.; Ahn, Y.H.; Park, E.J.; Jin, Y.H.; Jang, Y.K. JIB-04, A Small Molecule Histone Demethylase Inhibitor, Selectively Targets Colorectal Cancer Stem Cells by Inhibiting the Wnt/beta-Catenin Signaling Pathway. Sci. Rep. 2018, 8, 6611. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Feng, F.; Wu, J.; Fan, S.; Han, J.; Wang, S.; Yang, L.; Liu, W.; Wang, C.; Xu, K. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 2022, 181, 106270. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Tong, M. Protein Posttranslational Modification in Stemness Remodeling and Its Emerging Role as a Novel Therapeutic Target in Gastrointestinal Cancers. Int. J. Mol. Sci. 2023, 24, 9173. https://doi.org/10.3390/ijms24119173
Wang Y, Tong M. Protein Posttranslational Modification in Stemness Remodeling and Its Emerging Role as a Novel Therapeutic Target in Gastrointestinal Cancers. International Journal of Molecular Sciences. 2023; 24(11):9173. https://doi.org/10.3390/ijms24119173
Chicago/Turabian StyleWang, Yifei, and Man Tong. 2023. "Protein Posttranslational Modification in Stemness Remodeling and Its Emerging Role as a Novel Therapeutic Target in Gastrointestinal Cancers" International Journal of Molecular Sciences 24, no. 11: 9173. https://doi.org/10.3390/ijms24119173