
Homo cerevisiae—Leveraging Yeast for Investigating Protein–Protein Interactions and Their Role in Human Disease
 ,
, {kind=link}
{kind=link}
Abstract
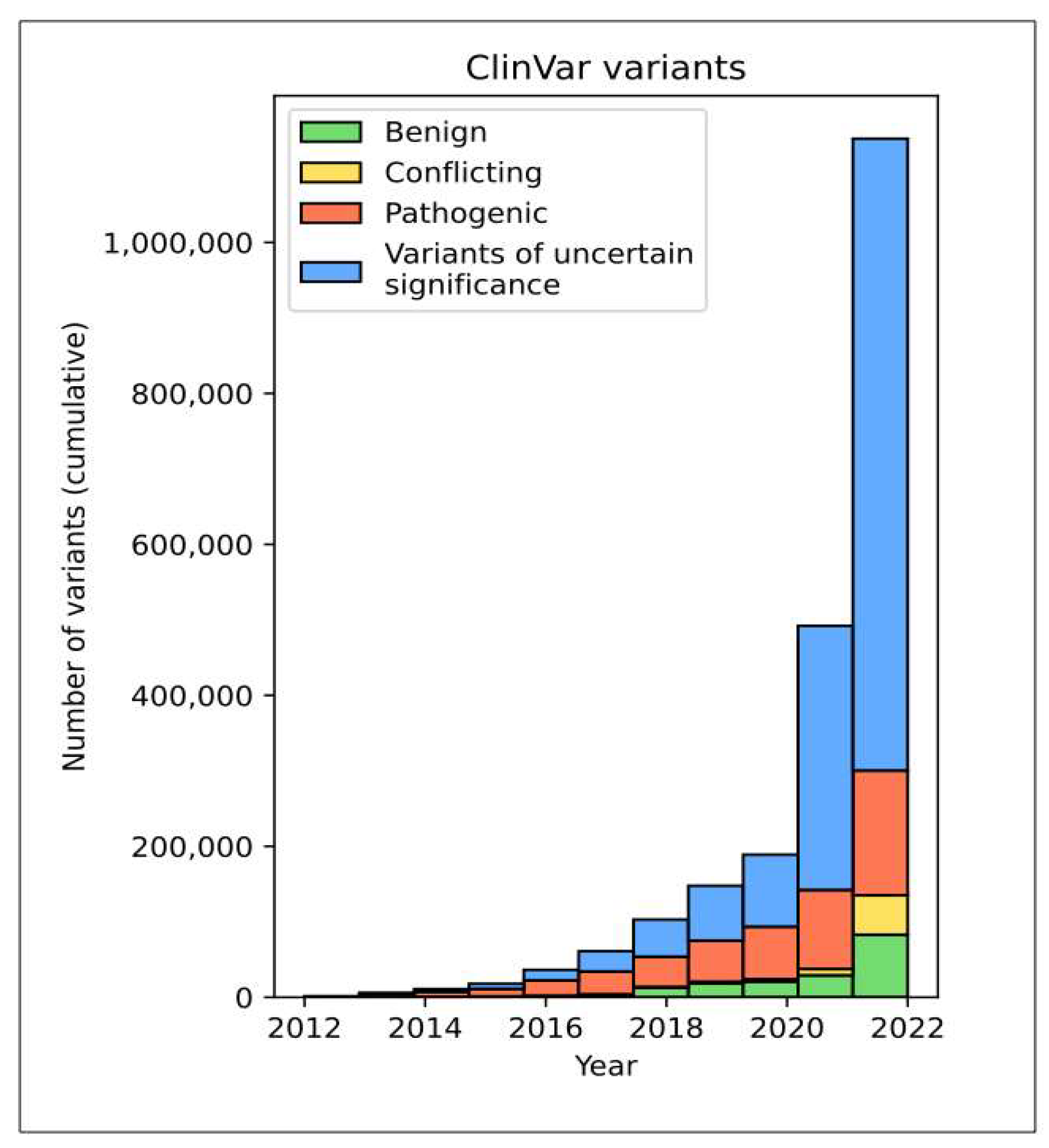
:1. The Disease-Causing Human Variome—The Curse of Too Much Knowledge
2. Edgetics—The Cutting Edge
3. Yeast, This Tiny but Mighty Organism
4. Yeast, or the Room of Requirement for Interactome Mapping
5. The Yeast Assayome—A Versatile Toolbox
6. Edgotyping of Genetic Variants—Action in the Interaction
7. Development of Therapies Targeting PPIs—The Usefulness of Useless Knowledge
8. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Samuels, M.E. Saturation of the Human Phenome. Curr. Genom. 2010, 11, 482–499. [Google Scholar] [CrossRef]
- Amberger, J.; Bocchini, C.; Hamosh, A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®). Hum. Mutat. 2011, 32, 564–567. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014, 43, D789–D798. [Google Scholar] [CrossRef]
- Amberger, J.S.; Hamosh, A. Searching Online Mendelian Inheritance in Man (OMIM): A Knowledgebase of Human Genes and Genetic Phenotypes. Curr. Protoc. Bioinform. 2017, 58, 1.2.1–1.2.12. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Howells, K.; Phillips, A.D.; Thomas, N.S.; Cooper, D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009, 1, 13. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2013, 133, 1–9. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Chakravarti, A.; Cohen, J.C.; Hardy, J. Mendelian disorders and multifactorial traits: The big divide or one for all. Nat. Rev. Genet. 2010, 11, 380–384. [Google Scholar] [CrossRef]
- Majewski, J.; Schwartzentruber, J.; LaLonde, E.; Montpetit, A.; Jabado, N. What can exome sequencing do for you. Nat. Rev. Genet. 2011, 48, 580–589. [Google Scholar] [CrossRef]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.E.; Othman, M.; Jakobsdottir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein–associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef]
- Neale, B.M.; Fagerness, J.; Reynolds, R.; Sobrin, L.; Parker, M.; Raychaudhuri, S.; Tan, P.L.; Oh, E.C.; Merriam, J.E.; Souied, E.; et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl. Acad. Sci. USA 2010, 107, 7395–7400. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, S.; Takahashi, A.; Ashikawa, K.; Hosono, N.; Aoi, T.; Yasuda, M.; Oshima, Y.; Yoshida, S.; Enaida, H.; Tsuchihashi, T.; et al. Genome-wide association study identifies two susceptibility loci for exudative age-related macular degeneration in the Japanese population. Nat. Genet. 2011, 43, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bhangale, T.R.; Fagerness, J.; Ripke, S.; Thorleifsson, G.; Tan, P.L.; Souied, E.H.; Richardson, A.J.; Merriam, J.E.; Buitendijk, G.H.; et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum. Mol. Genet. 2011, 20, 3699–3709. [Google Scholar] [CrossRef] [PubMed]
- Tryka, K.A.; Hao, L.; Sturcke, A.; Jin, Y.; Wang, Z.Y.; Ziyabari, L.; Lee, M.; Popova, N.; Sharopova, N.; Kimura, M.; et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2013, 42, D975–D979. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2016, 45, D896–D901. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- Huang, H.; Fang, M.; Jostins, L.; Mirkov, M.U.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013, 132, 1077–1130. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Zhong, Q.; Simonis, N.; Li, Q.-R.; Charloteaux, B.; Heuze, F.; Klitgord, N.; Tam, S.; Yu, H.; Venkatesan, K.; Mou, D.; et al. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 2009, 5, 321. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.S.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional Links Between Aβ Toxicity, Endocytic Trafficking, and Alzheimer’s Disease Risk Factors in Yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Olivet, J.; Maseko, S.B.; Volkov, A.N.; Salehi-Ashtiani, K.; Das, K.; Calderwood, M.A.; Twizere, J.-C.; Gorgulla, C. A systematic approach to identify host targets and rapidly deliver broad-spectrum antivirals. Mol. Ther. 2022, 30, 1797–1800. [Google Scholar] [CrossRef] [PubMed]
- Thali, M.; Olshevsky, U.; Furman, C.; Gabuzda, D.; Posner, M.; Sodroski, J. Characterization of a discontinuous human immunodeficiency virus type 1 gp120 epitope recognized by a broadly reactive neutralizing human monoclonal antibody. J. Virol. 1991, 65, 6188–6193. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef]
- Kim, D.-K.; Weller, B.; Lin, C.-W.; Sheykhkarimli, D.; Knapp, J.J.; Dugied, G.; Zanzoni, A.; Pons, C.; Tofaute, M.J.; Maseko, S.B.; et al. A proteome-scale map of the SARS-CoV-2–human contactome. Nat. Biotechnol. 2022, 41, 140–149. [Google Scholar] [CrossRef]
- Kachroo, A.H.; Vandeloo, M.; Greco, B.M.; Abdullah, M. Humanized yeast to model human biology, disease and evolution. Dis. Model. Mech. 2022, 15, dmm049309. [Google Scholar] [CrossRef]
- Hunter, P. The paradox of model organisms. The use of model organisms in research will continue despite their shortcomings. EMBO Rep. 2008, 9, 717–720. [Google Scholar] [CrossRef]
- Remm, M.; Storm, C.E.; Sonnhammer, E. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001, 314, 1041–1052. [Google Scholar] [CrossRef]
- Marcotte, E.M.; Pellegrini, M.; Ng, H.-L.; Rice, D.W.; Yeates, T.O.; Eisenberg, D. Detecting Protein Function and Protein-Protein Interactions from Genome Sequences. Science 1999, 285, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.E.; Suh, P.-G.; Kim, J.-I. O-GlcNAcylation in health and neurodegenerative diseases. Exp. Mol. Med. 2021, 53, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef]
- Sun, S.; Yang, F.; Tan, G.; Costanzo, M.; Oughtred, R.; Hirschman, J.; Theesfeld, C.L.; Bansal, P.; Sahni, N.; Yi, S.; et al. An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res. 2016, 26, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Fink, G.R. Yeast: An Experimental Organism for 21st Century Biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R.; Kolodner, R.D. Identification of mismatch repair genes and their role in the development of cancer. Curr. Opin. Genet. Dev. 1995, 5, 382–395. [Google Scholar] [CrossRef]
- Gasch, A.P.; Werner-Washburne, M. The genomics of yeast responses to environmental stress and starvation. Funct. Integr. Genom. 2002, 2, 181–192. [Google Scholar] [CrossRef]
- Kültz, D. Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef]
- Hartwell, L.H. Saccharomyces cerevisiae cell cycle. Bacteriol. Rev. 1974, 38, 164–198. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, G.J.C. Newborn screening in Latin America at the beginning of the 21st century. J. Inherit. Metab. Dis. 2007, 30, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic Control of the Cell Division Cycle in Yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, R.K.; Johnston, J.R. Life Span of Individual Yeast Cells. Nature 1959, 183, 1751–1752. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, P.; Longo, V.D. The chronological life span of Saccharomyces cerevisiae. Aging Cell 2003, 2, 73–81. [Google Scholar] [CrossRef]
- Pan, Y.; Shadel, G.S. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging 2009, 1, 131–145. [Google Scholar] [CrossRef]
- Mirisola, M.G.; Taormina, G.; Fabrizio, P.; Wei, M.; Hu, J.; Longo, V.D. Serine- and Threonine/Valine-Dependent Activation of PDK and Tor Orthologs Converge on Sch9 to Promote Aging. PLoS Genet. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Cyr, D.M. and Douglas, M.G. Differential regulation of Hsp70 subfamilies by the eukaryotic DnaJ homologue YDJ1. J. Biol. Chem. 1994, 269, 9798–9804. [Google Scholar] [CrossRef] [PubMed]
- Cajo, G.C.; Horne, B.E.; Kelley, W.L.; Schwager, F.; Georgopoulos, C.; Genevaux, P. The Role of the DIF Motif of the DnaJ (Hsp40) Co-chaperone in the Regulation of the DnaK (Hsp70) Chaperone Cycle. J. Biol. Chem. 2006, 281, 12436–12444. [Google Scholar] [CrossRef]
- Schatz, G. The isolation of possible mitochondrial precursor structures from aerobically grown baker’-s yeast. Biochem. Biophys. Res. Commun. 1963, 12, 448–451. [Google Scholar] [CrossRef]
- Foury, F.; Vanderstraeten, S. Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J. 1992, 11, 2717–2726. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Dieckmann, C.L. PET genes of Saccharomyces cerevisiae. Microbiol. Rev. 1990, 54, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gérard, B.; Rotig, A.; Saudubray, J.; Munnich, A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef]
- Pfanner, N.; van der Laan, M.; Amati, P.; Capaldi, R.A.; Caudy, A.A.; Chacinska, A.; Darshi, M.; Deckers, M.; Hoppins, S.; Icho, T.; et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014, 204, 1083–1086. [Google Scholar] [CrossRef]
- Cervelli, T.; Galli, A. Yeast as a Tool to Understand the Significance of Human Disease-Associated Gene Variants. Genes 2021, 12, 1303. [Google Scholar] [CrossRef]
- Ishioka, C.; Frebourg, T.; Yan, Y.-X.; Vidal, M.; Friend, S.H.; Schmidt, S.; Iggo, R. Screening patients for heterozygous p53 mutations using a functional assay in yeast. Nat. Genet. 1993, 5, 124–129. [Google Scholar] [CrossRef]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.Y.; Toufighi, K.; Mostafavi, S.; et al. The Genetic Landscape of a Cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef]
- Reddy, G.; Desai, M.M. Global epistasis emerges from a generic model of a complex trait. Elife 2021, 10, e64740. [Google Scholar] [CrossRef] [PubMed]
- Jerison, E.R.; Kryazhimskiy, S.; Mitchell, J.K.; Bloom, J.S.; Kruglyak, L.; Desai, M.M. Genetic variation in adaptability and pleiotropy in budding yeast. Elife 2017, 6, e27167. [Google Scholar] [CrossRef]
- Masison, D.C.; Reidy, M. Yeast prions are useful for studying protein chaperones and protein quality control. Prion 2015, 9, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Thouvenot, P.; Fourrière, L.; Dardillac, E.; Ben Yamin, B.; Lescure, A.; Lejour, V.; Heiligenstein, X.; Boulé, J.-B.; Romao, M.; Raposo-Benedetti, G.; et al. Yeast cells reveal the misfolding and the cellular mislocalisation of the human BRCA1 protein. J. Cell Sci. 2016, 129, 4366–4378. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.L.; Nyström, T.; Widlund, P.O. Studying Spatial Protein Quality Control, Proteopathies, and Aging Using Different Model Misfolding Proteins in S. cerevisiae. Front. Mol. Neurosci. 2018, 11, 249. [Google Scholar] [CrossRef]
- E Di Gregorio, S.; Duennwald, M.L. Yeast as a model to study protein misfolding in aged cells. FEMS Yeast Res. 2018, 18. [Google Scholar] [CrossRef]
- Samant, R.S.; Frydman, J. Methods for measuring misfolded protein clearance in the budding yeast Saccharomyces cerevisiae. Methods Enzymol. 2019, 619, 27–45. [Google Scholar] [CrossRef]
- Romero-Suarez, D.; Wulff, T.; Rong, Y.; Jakočiu̅nas, T.; Yuzawa, S.; Keasling, J.D.; Jensen, M.K. A Reporter System for Cytosolic Protein Aggregates in Yeast. ACS Synth. Biol. 2021, 10, 466–477. [Google Scholar] [CrossRef]
- Klein, J.; Heal, J.R.; Hamilton, W.D.O.; Boussemghoune, T.; Tange, T.; Delegrange, F.; Jaeschke, G.; Hatsch, A.; Heim, J. Yeast Synthetic Biology Platform Generates Novel Chemical Structures as Scaffolds for Drug Discovery. ACS Synth. Biol. 2014, 3, 314–323. [Google Scholar] [CrossRef]
- Romanos, M.A.; Scorer, C.A.; Clare, J.J. Foreign gene expression in yeast: A review. Yeast 1992, 8, 423–488. [Google Scholar] [CrossRef]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Keasling, J.D. Synergies between synthetic biology and metabolic engineering. Nat. Biotechnol. 2011, 29, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U. Systems strategies for developing industrial microbial strains. Nat. Biotechnol. 2015, 33, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.S.; Surette, M.G.; Elowitz, M.B. Programming gene expression with combinatorial promoters. Mol. Syst. Biol. 2007, 3, 145. [Google Scholar] [CrossRef]
- Daniel, R.; Rubens, J.R.; Sarpeshkar, R.; Lu, T.K. Synthetic analog computation in living cells. Nature 2013, 497, 619–623. [Google Scholar] [CrossRef]
- Nielsen, A.A.K.; Der, B.S.; Shin, J.; Vaidyanathan, P.; Paralanov, V.; Strychalski, E.A.; Ross, D.; Densmore, D.; Voigt, C.A. Genetic circuit design automation. Science 2016, 352, aac7341. [Google Scholar] [CrossRef]
- Hillenmeyer, M.E.; Fung, E.; Wildenhain, J.; Pierce, S.E.; Hoon, S.; Lee, W.; Proctor, M.; St.Onge, R.P.; Tyers, M.; Koller, D.; et al. The Chemical Genomic Portrait of Yeast: Uncovering a Phenotype for All Genes. Science 2008, 320, 362–365. [Google Scholar] [CrossRef]
- Olivet, J.; Choi, S.G.; Sierra, S.; O’Grady, T.M.; de la Fuente Revenga, M.; Laval, F.; Botchkarev, V.V., Jr.; Gorgulla, C.; Coote, P.W.; Blavier, J. Expanding the HDAC druggable landscape beyond enzymatic activity. bioRxiv 2023. [Google Scholar] [CrossRef]
- E Jackrel, M.; Shorter, J. Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 2015, 9, 90–109. [Google Scholar] [CrossRef]
- Fromont-Racine, M.; Rain, J.-C.; Legrain, P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet. 1997, 16, 277–282. [Google Scholar] [CrossRef]
- Walhout, A.J.M.; Sordella, R.; Lu, X.; Hartley, J.L.; Temple, G.F.; Brasch, M.A.; Thierry-Mieg, N.; Vidal, M. Protein Interaction Mapping in C. elegans Using Proteins Involved in Vulval Development. Science 2000, 287, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Chiba, T.; Ozawa, R.; Yoshida, M.; Hattori, M.; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar] [CrossRef] [PubMed]
- Reboul, J.; Vaglio, P.; Rual, J.-F.; Lamesch, P.; Martinez, M.; Armstrong, C.M.; Li, S.; Jacotot, L.; Bertin, N.; Janky, R.; et al. C. elegans ORFeome version 1.1: Experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat. Genet. 2003, 34, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Armstrong, C.M.; Bertin, N.; Ge, H.; Milstein, S.; Boxem, M.; Vidalain, P.-O.; Han, J.-D.J.; Chesneau, A.; Hao, T.; et al. A Map of the Interactome Network of the Metazoan C. elegans. Science 2004, 303, 540–543. [Google Scholar] [CrossRef]
- Arabidopsis Interactome Mapping Consortium; Dreze, M.; Carvunis, A.-R.; Charloteaux, B.; Galli, M.; Pevzner, S.J.; Tasan, M.; Ahn, Y.-Y.; Balumuri, P.; Barabási, A.-L.; et al. Evidence for Network Evolution in an Arabidopsis Interactome Map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef]
- Zhong, Q.; Pevzner, S.J.; Hao, T.; Wang, Y.; Mosca, R.; Menche, J.; Taipale, M.; Taşan, M.; Fan, C.; Yang, X.; et al. An inter-species protein–protein interaction network across vast evolutionary distance. Mol. Syst. Biol. 2016, 12, 865. [Google Scholar] [CrossRef]
- Tang, H.-W.; Spirohn, K.; Hu, Y.; Hao, T.; Kovács, I.A.; Gao, Y.; Binari, R.; Yang-Zhou, D.; Wan, K.H.; Bader, J.S.; et al. Next-generation large-scale binary protein interaction network for Drosophila melanogaster. Nat. Commun. 2023, 14, 2162. [Google Scholar] [CrossRef] [PubMed]
- Rual, J.-F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A Proteome-Scale Map of the Human Interactome Network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Willyard, C. New human gene tally reignites debate. Nature 2018, 558, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, M.A.; Venkatesan, K.; Xing, L.; Chase, M.R.; Vazquez, A.; Holthaus, A.M.; Ewence, A.E.; Li, N.; Hirozane-Kishikawa, T.; Hill, D.E.; et al. Epstein–Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA 2007, 104, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Vandermeulen, C.; O’grady, T.; Wayet, J.; Galvan, B.; Maseko, S.; Cherkaoui, M.; Desbuleux, A.; Coppin, G.; Olivet, J.; Ben Ameur, L.; et al. The HTLV-1 viral oncoproteins Tax and HBZ reprogram the cellular mRNA splicing landscape. PLoS Pathog. 2021, 17, e1009919. [Google Scholar] [CrossRef] [PubMed]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Pujana, M.A.; Han, J.-D.J.; Starita, L.M.; Stevens, K.N.; Tewari, M.; Ahn, J.S.; Rennert, G.; Moreno, V.; Kirchhoff, T.; Gold, B.; et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007, 39, 1338–1349. [Google Scholar] [CrossRef]
- Kahle, J.J.; Gulbahce, N.; Shaw, C.A.; Lim, J.; Hill, D.E.; Barabási, A.-L.; Zoghbi, H.Y. Comparison of an expanded ataxia interactome with patient medical records reveals a relationship between macular degeneration and ataxia. Hum. Mol. Genet. 2010, 20, 510–527. [Google Scholar] [CrossRef]
- Sakai, Y.; Shaw, C.A.; Dawson, B.C.; Dugas, D.V.; Al-Mohtaseb, Z.; Hill, D.E.; Zoghbi, H.Y. Protein Interactome Reveals Converging Molecular Pathways Among Autism Disorders. Sci. Transl. Med. 2011, 3, 86ra49. [Google Scholar] [CrossRef]
- Corominas, R.; Yang, X.; Lin, G.N.; Kang, S.; Shen, Y.; Ghamsari, L.; Broly, M.; Rodriguez, M.; Tam, S.; Trigg, S.A.; et al. Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nat. Commun. 2014, 5, 3650. [Google Scholar] [CrossRef]
- Choi, S.G.; Olivet, J.; Cassonnet, P.; Vidalain, P.-O.; Luck, K.; Lambourne, L.; Spirohn, K.; Lemmens, I.; Dos Santos, M.; Demeret, C.; et al. Maximizing binary interactome mapping with a minimal number of assays. Nat. Commun. 2019, 10, 3907. [Google Scholar] [CrossRef] [PubMed]
- Cafarelli, T.; Desbuleux, A.; Wang, Y.; Choi, S.; De Ridder, D.; Vidal, M. Mapping, modeling, and characterization of protein–protein interactions on a proteomic scale. Curr. Opin. Struct. Biol. 2017, 44, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O.-K. A novel genetic system to detect protein–protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A.J.; Boulton, S.J.; Vidal, M. Yeast two-hybrid systems and protein interaction mapping projects for yeast and worm. Yeast 2000, 17, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Fashena, S.J.; Serebriiskii, I.G.; Golemis, E.A. [2] LexA-based two-hybrid systems. Methods Enzymol. 2000, 328, 14–26. [Google Scholar] [CrossRef]
- Hopper, J.E.; Broach, J.R.; Rowe, L.B. Regulation of the galactose pathway in Saccharomyces cerevisiae: Induction of uridyl transferase mRNA and dependency on GAL4 gene function. Proc. Natl. Acad. Sci. USA 1978, 75, 2878–2882. [Google Scholar] [CrossRef]
- Bram, R.J.; Kornberg, R.D. Specific protein binding to far upstream activating sequences in polymerase II promoters. Proc. Natl. Acad. Sci. USA 1985, 82, 43–47. [Google Scholar] [CrossRef]
- Giniger, E.; Varnum, S.M.; Ptashne, M. Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 1985, 40, 767–774. [Google Scholar] [CrossRef]
- Keegan, L.; Gill, G.; Ptashne, M. Separation of DNA Binding from the Transcription-Activating Function of a Eukaryotic Regulatory Protein. Science 1986, 231, 699–704. [Google Scholar] [CrossRef]
- Vidal, M.; Brachmann, R.K.; Fattaey, A.; Harlow, E.; Boeke, J.D. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 10315–10320. [Google Scholar] [CrossRef] [PubMed]
- Van Criekinge, W.; Beyaert, R. Yeast two-hybrid: State of the art. Biol. Proced. Online 1999, 2, 1–38. [Google Scholar] [CrossRef]
- Heintz, V.J.; Wang, L.; LaCount, A.D.J. NanoLuc luciferase as a quantitative yeast two-hybrid reporter. FEMS Yeast Res. 2021, 21, foab069. [Google Scholar] [CrossRef] [PubMed]
- Yachie, N.; Petsalaki, E.; Mellor, J.C.; Weile, J.; Jacob, Y.; Verby, M.; Ozturk, S.B.; Li, S.; Cote, A.G.; Mosca, R.; et al. Pooled-matrix protein interaction screens using Barcode Fusion Genetics. Mol. Syst. Biol. 2016, 12, 863. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.-F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.-I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2008, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M. How much of the human protein interactome remains to be mapped. Sci. Signal. 2016, 9, eg7. [Google Scholar] [CrossRef]
- Fulmer, S.B.; Schwiebert, E.M.; Morales, M.M.; Guggino, W.B.; Cutting, G.R. Two cystic fibrosis transmembrane conductance regulator mutations have different effects on both pulmonary phenotype and regulation of outwardly rectified chloride currents. Proc. Natl. Acad. Sci. USA 1995, 92, 6832–6836. [Google Scholar] [CrossRef]
- Gilbert, A.; Jadot, M.; Leontieva, E.; Coninck, S.W.-D.; Wattiaux, R. ΔF508 CFTR Localizes in the Endoplasmic Reticulum–Golgi Intermediate Compartment in Cystic Fibrosis Cells. Exp. Cell Res. 1998, 242, 144–152. [Google Scholar] [CrossRef]
- Dérand, R.; Bulteau-Pignoux, L.; Becq, F. The Cystic Fibrosis Mutation G551D Alters the Non-Michaelis-Menten Behavior of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channel and Abolishes the Inhibitory Genistein Binding Site. J. Biol. Chem. 2002, 277, 35999–36004. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Harada, K.; Takeya, M.; Yamahira, K.; Wada, I.; Shuto, T.; Suico, M.A.; Hashimoto, Y.; Kai, H. ΔF508 CFTR Pool in the Endoplasmic Reticulum Is Increased by Calnexin Overexpression. Mol. Biol. Cell 2004, 15, 563–574. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Bunn, H.F.; Forget, B.F. Haemoglobin: Molecular, Genetic and Clinical Aspects, 2nd ed.; W.B. Saunders Co.: London, UK; Available online: https://agris.fao.org/agris-search/search.do?recordID=US201300647987 (accessed on 18 May 2023).
- Parikh, A.A.; Ellis, L.M. The vascular endothelial growth factor family and its receptors. Hematol. Clin. N. Am. 2004, 18, 951–971. [Google Scholar] [CrossRef] [PubMed]
- Serjeant, G.R. The Natural History of Sickle Cell Disease. Cold Spring Harb. Perspect. Med. 2013, 3, a011783. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Zhong, Q.; Jailkhani, N.; Charloteaux, B.; E Cusick, M.; Vidal, M. Edgotype: A fundamental link between genotype and phenotype. Curr. Opin. Genet. Dev. 2013, 23, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Taipale, M.; Bass, J.I.F.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread Macromolecular Interaction Perturbations in Human Genetic Disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef]
- Barshir, R.; Shwartz, O.; Smoly, I.Y.; Yeger-Lotem, E. Comparative Analysis of Human Tissue Interactomes Reveals Factors Leading to Tissue-Specific Manifestation of Hereditary Diseases. PLOS Comput. Biol. 2014, 10, e1003632. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Fry, D.C. Protein–protein interactions as targets for small molecule drug discovery. Biopolymers 2006, 84, 535–552. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]
- Dang, T.O.; Ogunniyi, A.; Barbee, M.S.; Drilon, A. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev. Anticancer. Ther. 2015, 16, 13–20. [Google Scholar] [CrossRef]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Panaccione, R.; Gordon, K.B.; McIlraith, M.J.; Lacerda, A.P.M. Adalimumab: Long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn’s disease. Ann. Rheum. Dis. 2012, 72, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Letai, A. ABT-199: Taking Dead Aim at BCL-2. Cancer Cell 2013, 23, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2002, 2, 364–371. [Google Scholar] [CrossRef]
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The HER-2 Receptor and Breast Cancer: Ten Years of Targeted Anti–HER-2 Therapy and Personalized Medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laval, F.; Coppin, G.; Twizere, J.-C.; Vidal, M. Homo cerevisiae—Leveraging Yeast for Investigating Protein–Protein Interactions and Their Role in Human Disease. Int. J. Mol. Sci. 2023, 24, 9179. https://doi.org/10.3390/ijms24119179
Laval F, Coppin G, Twizere J-C, Vidal M. Homo cerevisiae—Leveraging Yeast for Investigating Protein–Protein Interactions and Their Role in Human Disease. International Journal of Molecular Sciences. 2023; 24(11):9179. https://doi.org/10.3390/ijms24119179
Chicago/Turabian StyleLaval, Florent, Georges Coppin, Jean-Claude Twizere, and Marc Vidal. 2023. "Homo cerevisiae—Leveraging Yeast for Investigating Protein–Protein Interactions and Their Role in Human Disease" International Journal of Molecular Sciences 24, no. 11: 9179. https://doi.org/10.3390/ijms24119179