
Molecular, Electrophysiological, and Ultrasonographic Differences in Selected Immune-Mediated Neuropathies with Therapeutic Implications

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- distal CIDP (synonyms: distal acquired demyelinating symmetric neuropathy), 7–15% presents with sensory loss in the distal limbs as well as gait instability,
- multifocal CIDP (synonyms: multifocal acquired demyelinating sensory and motor neuropathy [MADSAM]; multifocal demyelinating neuropathy with persistent conduction block, Lewis–Sumner syndrome [LSS]; multifocal inflammatory demyelinating neuropathy), 4–14% involvement of asymmetric sensory and motor fibers, more often in the upper limbs,
- focal CIDP 4–14% involvement of one limb or nerve plexus (usually the brachial or lumbosacral plexus),
- sensory CIDP 3.5–14%—only sensory fibers are involved, characterized by gait ataxia, impairment of vibration and position sense and changes in cutaneous sensation,
2. Methods
2.1. The Diagnostic Methods
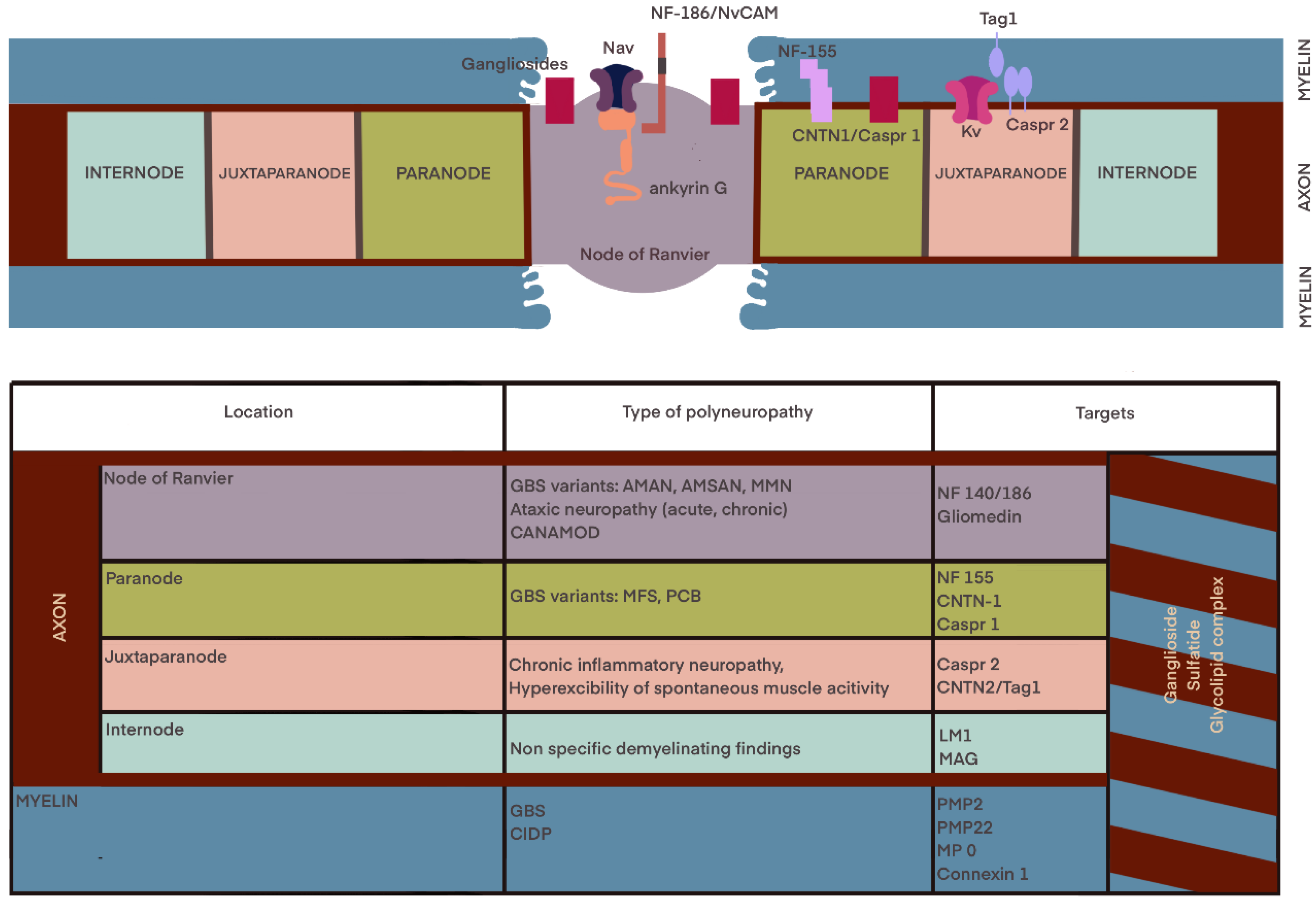
2.1.1. The Molecular Diagnostic
The Serum Biomarkers
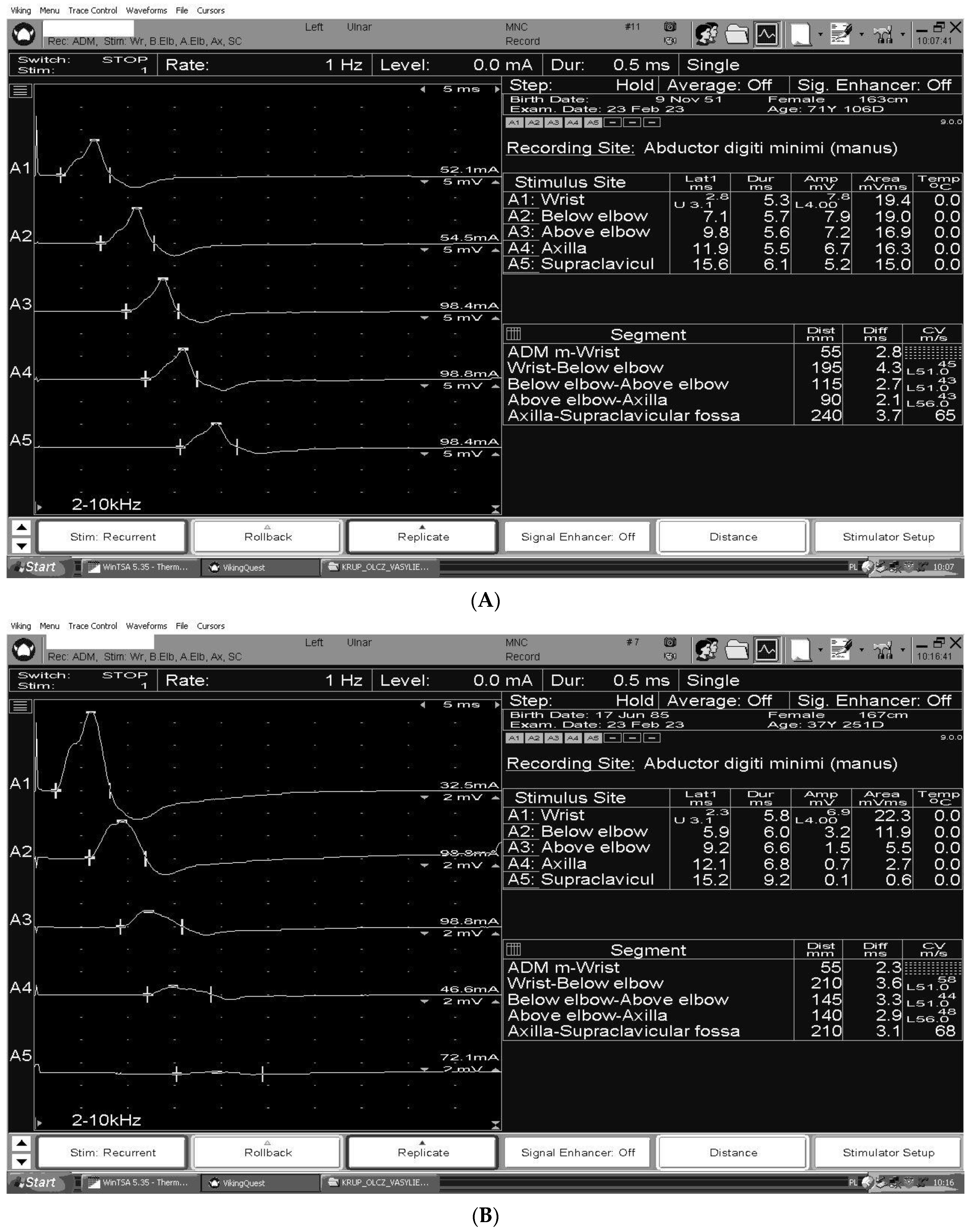
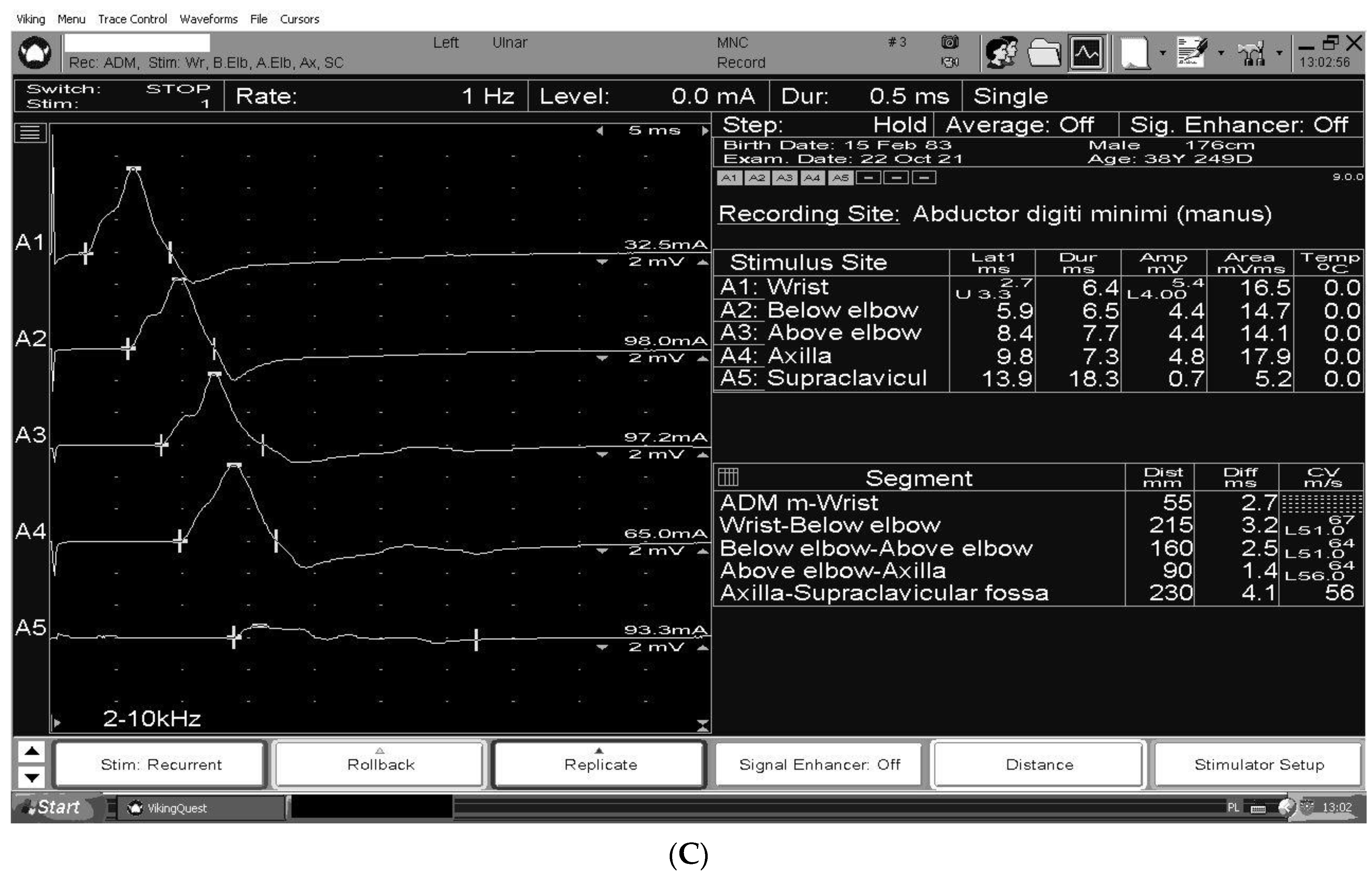
3. The Electrodiagnosis
4. Ultrasound
5. Treatment
Improved Clinical Criteria and the Development of More Disease-Specific
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMSAN | acute motor and sensory axonal neuropathy |
| AMAN | acute motor axonal neuropathy |
| GBS | Guillain-Barre syndrome |
| CNTN1 | cell adhesion molecules contactin 1 |
| CANOMAD | chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M [IgM] paraprotein, cold agglutinins, and disialosyl antibodies |
| CIDP | chronic inflammatory demyelinating polyneuropathy |
| CMAP | compound muscle action potential |
| Caspr1 | contactin-related protein 1 |
| CSA | cross-sectional area |
| IVIg | intravenous immunoglobulin |
| MFS | Miller Fisher Syndrome |
| MMN | multifocal motor neuropathy |
| MADSAM | multifocal aquired demyelinating sensory and motor neuropathy |
| NF155 | neurofascin 155 |
| NfL | neurofilament light chain |
| PE | plasma exchange |
| POEMS syndrome | Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal plasma cell disorder, Skin changes |
| LM1 | sialosylneolactotetraosylceramide glycolipid |
| SCIg | subcutaneous immunoglobulin |
| SGPG | sulfated glucuronosyl paragloboside |
References
- Gwathmey, K.G.; Smith, A.G. Immune-Mediated Neuropathies. Neurol. Clin. 2020, 38, 711–735. [Google Scholar] [CrossRef] [PubMed]
- Stino, A.M.; Naddaf, E.; Dyck, P.J.; Dyck, P.J.B. Chronic inflammatory demyelinating polyradiculoneuropathy-Diagnostic pitfalls and treatment approach. Muscle Nerve 2021, 63, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A. The Misdiagnosis of CIDP: A Review. Neurol. Ther. 2020, 9, 43–54. [Google Scholar] [CrossRef]
- Winter, N.; Grimm, A. Nerve Imaging, Electrodiagnostics, and Clinical Examination-Three Musketeers to Differentiate Polyneuropathies. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Querol, L.; Crabtree, M.; Herepath, M.; Priedane, E.; Viejo Viejo, I.; Agush, S.; Sommerer, P. Systematic literature review of burden of illness in chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neurol. 2021, 268, 3706–3716. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, X. Chronic Inflammatory Demyelinating Polyradiculoneuropathy in Association with Concomitant Diseases: Identification and Management. Front. Immunol. 2022, 13, 890142. [Google Scholar] [CrossRef]
- Van den Bergh, P.Y.K.; van Doorn, P.A.; Hadden, R.D.M.; Avau, B.; Vankrunkelsven, P.; Allen, J.A.; Attarian, S.; Blomkwist-Markens, P.H.; Cornblath, D.R.; Eftimov, F.; et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force—Second revision. Eur. J. Neurol. 2021, 28, 3556–3583. [Google Scholar] [CrossRef]
- Ayrignac, X.; Viala, K.; Koutlidis, R.M.; Taïeb, G.; Stojkovic, T.; Musset, L.; Léger, J.M.; Fournier, E.; Maisonobe, T.; Bouche, P. Sensory chronic inflammatory demyelinating polyneuropathy: An under-recognized entity? Muscle Nerve 2013, 48, 727–732. [Google Scholar] [CrossRef]
- Van den Berg-Vos, R.M.; Van den Berg, L.H.; Franssen, H.; Vermeulen, M.; Witkamp, T.D.; Jansen, G.H.; Van Es, H.W.; Kerkhoff, H.; Wokke, J.H.J. Multifocal inflammatory demyelinating neuropathy: A distinct clinical entity? Neurology 2000, 54, 26–32. [Google Scholar] [CrossRef]
- Doneddu, P.E.; De Lorenzo, A.; Manganelli, F.; Cocito, D.; Fazio, R.; Briani, C.; Mazzeo, A.; Filosto, M.; Cosentino, G.; Benedetti, L.; et al. Comparison of the diagnostic accuracy of the 2021 EAN/PNS and 2010 EFNS/PNS diagnostic criteria for chronic inflammatory demyelinating polyradiculoneuropathy. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1239–1246. [Google Scholar] [CrossRef]
- European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Socie. J. Peripher. Nerv. Syst. 2010, 15, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Broers, M.C.; Bunschoten, C.; Drenthen, J.; Beck, T.A.O.; Brusse, E.; Lingsma, H.F.; Allen, J.A.; Lewis, R.A.; Doorn, P.A.; Jacobs, B.C. Misdiagnosis and diagnostic pitfalls of chronic inflammatory demyelinating polyradiculoneuropathy. Eur. J. Neurol. 2021, 28, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Gorson, K.C.; Gelinas, D. Challenges in the diagnosis of chronic inflammatory demyelinating polyneuropathy. Brain Behav. 2018, 8, e00932. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, U.J.; Rajabally, Y.A. Underdiagnosis and diagnostic delay in chronic inflammatory demyelinating polyneuropathy. J. Neurol. 2021, 268, 1366–1373. [Google Scholar] [CrossRef]
- Briani, C.; Visentin, A. Therapeutic Monoclonal Antibody Therapies in Chronic Autoimmune Demyelinating Neuropathies. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Steck, A.J. Anti-MAG neuropathy: From biology to clinical management. J. Neuroimmunol. 2021, 361, 577725. [Google Scholar] [CrossRef]
- Yasuda, H.; Tomizawa, Y.; Harada, S.; Sasaki, M.; Komatsu, N.; Ando, J.; Hattori, N.; Ando, M. Anti-myelin-associated-glycoprotein neuropathy successfully treated with tirabrutinib. Heliyon 2022, 8, e10928. [Google Scholar] [CrossRef]
- Dispenzieri, A. POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2019, 94, 812–827. [Google Scholar] [CrossRef]
- Brown, R.; Ginsberg, L. POEMS syndrome: Clinical update. J. Neurol. 2019, 266, 268–277. [Google Scholar] [CrossRef]
- Khouri, J.; Nakashima, M.; Wong, S. Update on the Diagnosis and Treatment of POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Gammopathy, and Skin Changes) Syndrome: A Review. JAMA Oncol. 2021, 7, 1383–1391. [Google Scholar] [CrossRef]
- Oudeman, J.; Eftimov, F.; Strijkers, G.J.; Schneiders, J.J.; Roosendaal, S.D.; Engbersen, M.P.; Froeling, M.; Goedee, H.S.; Van Doorn, P.A.; Caan, M.W.; et al. Diagnostic accuracy of MRI and ultrasound in chronic immune-mediated neuropathies. Neurology 2020, 94, e62–e74. [Google Scholar] [CrossRef]
- Csurhes, P.A.; Sullivan, A.-A.; Green, K.; Pender, M.P.; McCombe, P.A. T cell reactivity to P0, P2, PMP-22, and myelin basic protein in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Klehmet, J.; Goehler, J.; Ulm, L.; Kohler, S.; Meisel, C.; Meisel, A.; Harms, H. Effective treatment with intravenous immunoglobulins reduces autoreactive T-cell response in patients with CIDP. J. Neurol. Neurosurg. Psychiatry 2015, 86, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Hagen, K.M.; Ousman, S.S. The immune response and aging in chronic inflammatory demyelinating polyradiculoneuropathy. J. Neuroinflamm. 2021, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Staudt, M.; Diederich, J.M.; Meisel, C.; Meisel, A.; Klehmet, J. Differences in peripheral myelin antigen-specific T cell responses and T memory subsets in atypical versus typical CIDP. BMC Neurol. 2017, 17, 81. [Google Scholar] [CrossRef]
- Koike, H.; Ikeda, S.; Fukami, Y.; Nishi, R.; Kawagashira, Y.; Iijima, M.; Nakamura, T.; Kuwahara, M.; Kusunoki, S.; Katsuno, M.; et al. Complement deposition and macrophage-induced demyelination in CIDP with anti-LM1 antibodies. J. Neurol. Sci. 2020, 408, 116509. [Google Scholar] [CrossRef]
- Eftimov, F.; Lucke, I.M.; Querol, L.A.; Rajabally, Y.A.; Verhamme, C. Diagnostic challenges in chronic inflammatory demyelinating polyradiculoneuropathy. Brain 2021, 143, 3214–3224. [Google Scholar] [CrossRef]
- Devaux, J.J.; Miura, Y.; Fukami, Y.; Inoue, T.; Manso, C.; Belghazi, M.; Sekiguchi, K.; Kokubun, N.; Ichikawa, H.; Wong, A.H.Y.; et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016, 86, 800–807. [Google Scholar] [CrossRef]
- Khadilkar, S.V.; Kamat, S.; Patel, R. Nodo-paranodopathies: Concepts, Clinical Implications, and Management. Ann. Indian Acad. Neurol. 2022, 25, 1001–1008. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Taylor, R.S. Guillain Barre Syndrome; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Donofrio, P.D. Guillain-Barré Syndrome. CONTINUUM Lifelong Learn. Neurol. 2017, 23, 1295–1309. [Google Scholar] [CrossRef]
- Rees, J.H.; Soudain, S.E.; Gregson, N.A.; Hughes, R.A. Campylobacter jejuni infection and Guillain-Barré syndrome. N. Engl. J. Med. 1995, 333, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, M.M.; Lastovica, A.J.; Moran, A.P. Lipopolysaccharides from Campylobacter jejuni O:41 strains associated with Guillain-Barré syndrome exhibit mimicry of GM1 ganglioside. Infect. Immun. 1998, 66, 3649–3655. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N.; Taki, T.; Inagaki, F.; Kasama, T.; Takahashi, M.; Saito, K.; Handa, S.; Miyatake, T. A bacterium lipopolysaccharide that elicits Guillain-Barré syndrome has a GM1 ganglioside-like structure. J. Exp. Med. 1993, 178, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Moyano, A.L.; Comín, R.; Lardone, R.D.; Alaniz, M.E.; Theaux, R.; Irazoqui, F.J.; Nores, G.A. Validation of a rabbit model of neuropathy induced by immunization with gangliosides. J. Neurol. Sci. 2008, 272, 110–114. [Google Scholar] [CrossRef]
- Goodfellow, J.A.; Bowes, T.; Sheikh, K.; Odaka, M.; Halstead, S.K.; Humphreys, P.D.; Wagner, E.R.; Yuki, N.; Furukawa, K.; Furukawa, K.; et al. Overexpression of GD1a ganglioside sensitizes motor nerve terminals to anti-GD1a antibody-mediated injury in a model of acute motor axonal neuropathy. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1620–1628. [Google Scholar] [CrossRef]
- Campbell, C.I.; McGonigal, R.; Barrie, J.A.; Delaere, J.; Bracke, L.; Cunningham, M.E.; Yao, D.; Delahaye, T.; Van de Walle, I.; Willison, H.J. Complement inhibition prevents glial nodal membrane injury in a GM1 antibody-mediated mouse model. Brain Commun. 2022, 4, fcac306. [Google Scholar] [CrossRef]
- Kusunoki, S. Antibodies to Glycolipids in Guillain-Barré Syndrome, Miller Fisher Syndrome and Related Autoimmune Neurological Diseases. Adv. Neurobiol. 2023, 29, 479–495. [Google Scholar]
- Kaida, K.; Ariga, T.; Yu, R.K. Antiganglioside antibodies and their pathophysiological effects on Guillain-Barré syndrome and related disorders—A review. Glycobiology 2009, 19, 676–692. [Google Scholar] [CrossRef]
- Murakami, K.; Kajimoto, Y.; Ito, H. Acute Oropharyngeal Palsy Following Bilateral Adie’s Tonic Pupils Associated with Anti-GT1a and GQ1b IgG Antibodies. Intern. Med. 2022, 61, 3121–3124. [Google Scholar] [CrossRef]
- Roggenbuck, D.; Delmont, E.; Reinhold, D.; Schierack, P.; Conrad, K.; Boucraut, J. Autoimmune peripheral neuropathies and contribution of antiganglioside/sulphatide autoantibody testing. Mediterr. J. Rheumatol. 2020, 31, 10–18. [Google Scholar] [CrossRef]
- McGonigal, R.; Willison, H.J. The role of gangliosides in the organisation of the node of Ranvier examined in glycosyltransferase transgenic mice. J. Anat. 2022, 241, 1259–1271. [Google Scholar] [CrossRef]
- Giannotta, C.; Di Pietro, D.; Gallia, F.; Nobile-Orazio, E. Anti-sulfatide IgM antibodies in peripheral neuropathy: To test or not to test? Eur. J. Neurol. 2015, 22, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Nutma, E.; Marzin, M.; Puentes, F. Imaging immunological processes from blood to brain in amyotrophic lateral sclerosis. Clin. Exp. Immunol. 2021, 206, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Hagen, K.M.; Ousman, S.S. The Neuroimmunology of Guillain-Barré Syndrome and the Potential Role of an Aging Immune System. Front. Aging Neurosci. 2020, 12, 613628. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Triggers of Guillain-Barré Syndrome: Campylobacter jejuni Predominates. Int. J. Mol. Sci. 2022, 23, 14222. [Google Scholar] [CrossRef] [PubMed]
- Susuki, K.; Yuki, N.; Schafer, D.P.; Hirata, K.; Zhang, G.; Funakoshi, K.; Rasband, M.N. Dysfunction of nodes of Ranvier: A mechanism for anti-ganglioside antibody-mediated neuropathies. Exp. Neurol. 2012, 233, 534–542. [Google Scholar] [CrossRef]
- Yeh, W.Z.; Dyck, P.J.; van den Berg, L.H.; Kiernan, M.C.; Taylor, B.V. Multifocal motor neuropathy: Controversies and priorities. J. Neurol. Neurosurg. Psychiatry 2020, 91, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Beadon, K.; Guimarães-Costa, R.; Léger, J.-M. Multifocal motor neuropathy. Curr. Opin. Neurol. 2018, 31, 559–564. [Google Scholar] [CrossRef]
- Tozza, S.; Spina, E.; Iovino, A.; Iodice, R.; Dubbioso, R.; Ruggiero, L.; Nolano, M.; Manganelli, F. Value of Antibody Determinations in Chronic Dysimmune Neuropathies. Brain Sci. 2022, 13, 37. [Google Scholar] [CrossRef]
- Hameed, S.; Cascella, M. Multifocal Motor Neuropathy; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Léger, J.-M.; Guimarães-Costa, R.; Iancu Ferfoglia, R. The pathogenesis of multifocal motor neuropathy and an update on current management options. Ther. Adv. Neurol. Disord. 2015, 8, 109–122. [Google Scholar] [CrossRef]
- Susuki, K.; Rasband, M.N.; Tohyama, K.; Koibuchi, K.; Okamoto, S.; Funakoshi, K.; Hirata, K.; Baba, H.; Yuki, N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J. Neurosci. 2007, 27, 3956–3967. [Google Scholar] [CrossRef] [PubMed]
- Bocci, S.; Giannini, F.; Volpi, N.; Rossi, A.; Ginanneschi, F. Multifocal motor neuropathy occurring after acute motor axonal neuropathy: Two stages of the same disease? Neurol. Sci. 2022, 43, 1463–1465. [Google Scholar] [CrossRef]
- Yuki, N.; Watanabe, H.; Nakajima, T.; Späth, P.J. IVIG blocks complement deposition mediated by anti-GM1 antibodies in multifocal motor neuropathy. J. Neurol. Neurosurg. Psychiatry 2011, 82, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Roggenbuck, J.J.; Boucraut, J.; Delmont, E.; Conrad, K.; Roggenbuck, D. Diagnostic insights into chronic-inflammatory demyelinating polyneuropathies. Ann. Transl. Med. 2018, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Pathogenesis of immune-mediated neuropathies. Biochim. Biophys. Acta 2015, 1852, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Susuki, K. Node of Ranvier disruption as a cause of neurological diseases. ASN Neuro 2013, 5, 209–219. [Google Scholar] [CrossRef]
- Kmezic, I.; Samuelsson, K.; Finn, A.; Upate, Z.; Blennow, K.; Zetterberg, H.; Press, R. Neurofilament light chain and total tau in the differential diagnosis and prognostic evaluation of acute and chronic inflammatory polyneuropathies. Eur. J. Neurol. 2022, 29, 2810–2822. [Google Scholar] [CrossRef]
- García-Fernández, P.; Höfflin, K.; Rausch, A.; Strommer, K.; Neumann, A.; Cebulla, N.; Reinhold, A.-K.; Rittner, H.; Üçeyler, N.; Sommer, C. Systemic inflammatory markers in patients with polyneuropathies. Front. Immunol. 2023, 14, 1067714. [Google Scholar] [CrossRef]
- Illes, Z.; Blaabjerg, M. Chapter 9-Cerebrospinal fluid findings in Guillain–Barré syndrome and chronic inflammatory demyelinating polyneuropathies. In Cerebrospinal Fluid in Neurologic Disorders; Deisenhammer, F., Teunissen, C.E., Tumani HBT-H of CN, Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 125–138. [Google Scholar]
- Doneddu, P.E.; Cocito, D.; Manganelli, F.; Fazio, R.; Briani, C.; Filosto, M.; Benedetti, L.; Mazzeo, A.; Marfia, G.A.; Cortese, A.; et al. Atypical CIDP: Diagnostic criteria, progression and treatment response. Data from the Italian CIDP Database. J. Neurol. Neurosurg. Psychiatry 2019, 90, 125–132. [Google Scholar] [CrossRef]
- Verschueren, A.; Azulay, J.P.; Attarian, S.; Boucraut, J.; Pellissier, J.F.; Pouget, J. Lewis-Sumner syndrome and multifocal motor neuropathy. Muscle Nerve 2005, 31, 88–94. [Google Scholar] [CrossRef]
- Hosokawa, T.; Nakajima, H.; Unoda, K.; Yamane, K.; Doi, Y.; Ishida, S.; Kimura, F.; Hanafusa, T. An electrophysiological classification associated with Guillain-Barré syndrome outcomes. J. Neurol. 2014, 261, 1986–1993. [Google Scholar] [CrossRef]
- Uncini, A.; Kuwabara, S. The electrodiagnosis of Guillain-Barré syndrome subtypes: Where do we stand? Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2018, 129, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Pasutharnchat, N.; Ratanasirisawad, V.; Santananukarn, M.; Taychargumpoo, C.; Amornvit, J.; Chunharas, C. Sural-sparing pattern: A study against electrodiagnostic subtypes of Guillain-Barre syndrome. Clin. Neurophysiol. Pract. 2022, 7, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Moshe-Lilie, O.; Ensrud, E.; Ragole, T.; Nizar, C.; Dimitrova, D.; Karam, C. CIDP mimics: A case series. BMC Neurol. 2021, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.; Russo, M.; Rodolico, C.; Arimatea, I.; Vita, G.; Toscano, A.; Mazzeo, A. Long-term treatment with subcutaneous immunoglobulin in multifocal motor neuropathy. Sci. Rep. 2021, 11, 9216. [Google Scholar] [CrossRef]
- Padua, L.; Granata, G.; Sabatelli, M.; Inghilleri, M.; Lucchetta, M.; Luigetti, M.; Luigetti, M.; Coraci, D.; Martinoli, C.; Briani, C. Heterogeneity of root and nerve ultrasound pattern in CIDP patients. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2014, 125, 160–165. [Google Scholar] [CrossRef]
- Allen, J.A.; Lewis, R.A. CIDP diagnostic pitfalls and perception of treatment benefit. Neurology 2015, 85, 498–504. [Google Scholar] [CrossRef]
- Won, S.J.; Kim, B.-J.; Park, K.S.; Kim, S.H.; Yoon, J.S. Measurement of cross-sectional area of cervical roots and brachial plexus trunks. Muscle Nerve 2012, 46, 711–716. [Google Scholar] [CrossRef]
- Yalcin, E.; Onder, B.; Akyuz, M. Ulnar nerve measurements in healthy individuals to obtain reference values. Rheumatol. Int. 2013, 33, 1143–1147. [Google Scholar] [CrossRef]
- Grimm, A.; Heiling, B.; Schumacher, U.; Witte, O.W.; Axer, H. Ultrasound differentiation of axonal and demyelinating neuropathies. Muscle Nerve 2014, 50, 976–983. [Google Scholar] [CrossRef]
- Zaidman, C.M.; Harms, M.B.; Pestronk, A. Ultrasound of inherited vs. acquired demyelinating polyneuropathies. J. Neurol. 2013, 260, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Fisse, A.L.; Pitarokoili, K.; Trampe, N.; Motte, J.; Kerasnoudis, A.; Gold, R.; Yoon, M.-S. Clinical, Sonographic, and Electrophysiologic Longitudinal Features of Chronic Inflammatory Demyelinating Polyneuropathy. J. Neuroimaging 2019, 29, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kerasnoudis, A.; Pitarokoili, K.; Gold, R.; Yoon, M.-S. Nerve Ultrasound and Electrophysiology for Therapy Monitoring in Chronic Inflammatory Demyelinating Polyneuropathy. J. Neuroimaging 2015, 25, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Merola, A.; Rosso, M.; Romagnolo, A.; Peci, E.; Cocito, D. Peripheral Nerve Ultrasonography in Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Multifocal Motor Neuropathy: Correlations with Clinical and Neurophysiological Data. Neurol. Res. Int. 2016, 2016, 9478593. [Google Scholar] [CrossRef]
- Niu, J.; Ding, Q.; Fan, J.; Zhang, L.; Liu, J.; Guan, Y.; Wu, S.; Cui, L.; Liu, M. Nerve Ultrasound Performances in Differentiating POEMS Syndrome from CIDP. Neurotherapeutics 2022, 19, 455–463. [Google Scholar] [CrossRef]
- Kerasnoudis, A.; Pitarokoili, K.; Gold, R.; Yoon, M.-S. Bochum ultrasound score allows distinction of chronic inflammatory from multifocal acquired demyelinating polyneuropathies. J. Neurol. Sci. 2015, 348, 211–215. [Google Scholar] [CrossRef]
- Sugimoto, T.; Ochi, K.; Hosomi, N.; Takahashi, T.; Ueno, H.; Nakamura, T.; Nagano, Y.; Maruyama, H.; Kohriyama, T.; Matsumoto, M. Ultrasonographic nerve enlargement of the median and ulnar nerves and the cervical nerve roots in patients with demyelinating Charcot–Marie–Tooth disease: Distinction from patients with chronic inflammatory demyelinating polyneuropathy. J. Neurol. 2013, 260, 2580–2587. [Google Scholar] [CrossRef]
- Goedee, H.S.; van der Pol, W.L.; van Asseldonk, J.-T.H.; Franssen, H.; Notermans, N.C.; Vrancken, A.J.; van Es, M.A.; Nikolakopoulos, S.; Visser, L.H.; Berg, L.H.V.D. Diagnostic value of sonography in treatment-naive chronic inflammatory neuropathies. Neurology 2017, 88, 143–151. [Google Scholar] [CrossRef]
- Shah, S.; Chandrashekar, H.; Manji, H.; Davagnanam, I. Cranial nerve, spinal root and plexus hypertrophy in chronic inflammatory demyelinating polyneuropathy. Pract. Neurol. 2012, 12, 68–69. [Google Scholar] [CrossRef]
- Goedee, H.S.; Jongbloed, B.A.; van Asseldonk, J.-T.H.; Hendrikse, J.; Vrancken, A.F.J.E.; Franssen, H.; Nikolakopoulos, S.; Visser, L.H.; Pol, W.L.; Berg, L.H. A comparative study of brachial plexus sonography and magnetic resonance imaging in chronic inflammatory demyelinating neuropathy and multifocal motor neuropathy. Eur. J. Neurol. 2017, 24, 1307–1313. [Google Scholar] [CrossRef]
- Herraets, I.J.T.; Goedee, H.S.; Telleman, J.A.; van Eijk, R.P.A.; Verhamme, C.; Saris, C.G.J.; Eftimov, F.; van Alfen, N.; van Asseldonk, J.T.; Visser, L.H.; et al. Nerve ultrasound for diagnosing chronic inflammatory neuropathy: A multicenter validation study. Neurology 2020, 95, e1745–e1753. [Google Scholar] [CrossRef] [PubMed]
- Zaidman, C.M.; Al-Lozi, M.; Pestronk, A. Peripheral nerve size in normals and patients with polyneuropathy: An ultrasound study. Muscle Nerve 2009, 40, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Rattay, T.W.; Winter, N.; Décard, B.F.; Dammeier, N.M.; Härtig, F.; Ceanga, M.; Axer, H.; Grimm, A. Nerve ultrasound as follow-up tool in treated multifocal motor neuropathy. Eur. J. Neurol. 2017, 24, 1125–1134. [Google Scholar] [CrossRef]
- Décard, B.F.; Pham, M.; Grimm, A. Ultrasound and MRI of nerves for monitoring disease activity and treatment effects in chronic dysimmune neuropathies—Current concepts and future directions. Clin. Neurophysiol. 2018, 129, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Kerasnoudis, A.; Pitarokoili, K.; Behrendt, V.; Gold, R.; Yoon, M.-S. Nerve ultrasound score in distinguishing chronic from acute inflammatory demyelinating polyneuropathy. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2014, 125, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; Oki, R.; Shimizu, S.; Iijima, M.; Fukami, Y.; Tamura, N.; Nakatochi, M.; Ando, M.; Nishi, R.; Koike, H.; et al. Efficacy and Safety of Rituximab in Refractory CIDP with or without IgG4 Autoantibodies (RECIPE): Protocol for a Double-Blind, Randomized, Placebo-Controlled Clinical Trial. JMIR Res. Protoc. 2020, 9, e17117. [Google Scholar]
- Allen, J.A.; Gelinas, D.F.; Freimer, M.; Runken, M.C.; Wolfe, G.I. Immunoglobulin administration for the treatment of CIDP: IVIG or SCIG? J. Neurol. Sci. 2020, 408, 116497. [Google Scholar] [CrossRef]
- Goyal, N.A.; Karam, C.; Sheikh, K.A.; Dimachkie, M.M. Subcutaneous immunoglobulin treatment for chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 2021, 64, 243–254. [Google Scholar] [CrossRef]
- Urdiales-Sánchez, S.; González-Montaña, J.-R.; Diaz-Pérez, R.; Calvo-Calleja, P.; Gutiérrez-Trueba, M.-A.; Urdiales-Urdiales, J. Nodopathies in the Early Diagnosis of Axonal Forms of Guillain-Barré Syndrome. Front. Neurol. 2022, 13, 902172. [Google Scholar] [CrossRef]
- Berciano, J. Axonal pathology in early stages of Guillain-Barré syndrome. Neurologia 2022, 37, 466–479. [Google Scholar] [CrossRef]
- Tonev, D.G.; Momchilova, A.B. Therapeutic Plasma Exchange in Certain Immune-Mediated Neurological Disorders: Focus on a Novel Nanomembrane-Based Technology. Biomedicines 2023, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Berciano, J.; Sedano, M.J.; Pelayo-Negro, A.L.; García, A.; Orizaola, P.; Gallardo, E.; Lafarga, M.; Berciano, M.T.; Jacobs, B.C. Proximal nerve lesions in early Guillain-Barré syndrome: Implications for pathogenesis and disease classification. J. Neurol. 2017, 264, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Kokubun, N.; Nishibayashi, M.; Uncini, A.; Odaka, M.; Hirata, K.; Yuki, N. Conduction block in acute motor axonal neuropathy. Brain 2010, 133, 2897–2908. [Google Scholar] [CrossRef]
- Chevret, S.; Hughes, R.A.; Annane, D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2017, 2, CD001798. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.A.C.; Swan, A.V.; van Doorn, P.A. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2014, 2014, CD002063. [Google Scholar] [CrossRef] [PubMed]
- Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet 1997, 349, 225–230. [Google Scholar] [CrossRef]
- Walgaard, C.; Jacobs, B.C.; Lingsma, H.F.; Steyerberg, E.W.; van den Berg, B.; Doets, A.Y.; Leonhard, S.E.; Verboon, C.; Huizinga, R.; Drenthen, J.; et al. Second intravenous immunoglobulin dose in patients with Guillain-Barré syndrome with poor prognosis (SID-GBS): A double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2021, 20, 275–283. [Google Scholar] [CrossRef]
- Lin, J.; Gao, Q.; Xiao, K.; Tian, D.; Hu, W.; Han, Z. Efficacy of therapies in the treatment of Guillain-Barre syndrome: A network meta-analysis. Medicine 2021, 100, e27351. [Google Scholar] [CrossRef]
- Hughes, R.A.C.; Swan, A.V.; van Koningsveld, R.; van Doorn, P.A. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2006, 10, CD001446. [Google Scholar] [CrossRef]
- Hughes, R.A.; Brassington, R.; Gunn, A.A.; van Doorn, P.A. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2016, 10, CD001446. [Google Scholar] [CrossRef]
- Keddie, S.; Eftimov, F.; van den Berg, L.H.; Brassington, R.; de Haan, R.J.; van Schaik, I.N. Immunoglobulin for multifocal motor neuropathy. Cochrane Database Syst. Rev. 2022, 1, CD004429. [Google Scholar] [PubMed]
- Heo, Y.-A. Efgartigimod: First Approval. Drugs 2022, 82, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Spaeth, P.J. The importance of FcRn in neuro-immunotherapies: From IgG catabolism, FCGRT gene polymorphisms, IVIg dosing and efficiency to specific FcRn inhibitors. Ther. Adv. Neurol. Disord. 2021, 14, 1756286421997381. [Google Scholar] [CrossRef] [PubMed]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Pritchard, J.; Gray, I.A.; Idrissova, Z.R.; Lecky, B.R.F.; Sutton, I.J.; Swan, A.V.; Willison, H.J.; Winer, J.B.; Hughes, R.A. A randomized controlled trial of recombinant interferon-beta 1a in Guillain-Barré syndrome. Neurology 2003, 61, 1282–1284. [Google Scholar] [CrossRef]
- Bensa, S.; Hadden, R.D.; Hahn, A.; Hughes, R.A.; Willison, H.J. Randomized controlled trial of brain-derived neurotrophic factor in Guillain-Barré syndrome: A pilot study. Eur. J. Neurol. 2000, 7, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Wollinsky, K.H.; Hülser, P.J.; Brinkmeier, H.; Aulkemeyer, P.; Bössenecker, W.; Huber-Hartmann, K.H.; Rohrbach, P.; Schreiber, H.; Weber, F.; Kron, M.; et al. CSF filtration is an effective treatment of Guillain-Barré syndrome: A randomized clinical trial. Neurology 2001, 57, 774–780. [Google Scholar] [CrossRef]
- Zhang, X.; Xia, J.; Ye, H. Effect of Tripterygium polyglycoside on interleukin-6 in patients with Guillain-Barre syndrome. Chin. J. Integr. Tradit. West. Med. 2000, 20, 332–334. [Google Scholar]
- Fitzpatrick, A.M.; Mann, C.A.; Barry, S.; Brennan, K.; Overell, J.R.; Willison, H.J. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J. Peripher. Nerv. Syst. 2011, 16, 84–91. [Google Scholar] [CrossRef]
- Budding, K.; Johansen, L.E.; Van de Walle, I.; Dijkxhoorn, K.; de Zeeuw, E.; Bloemenkamp, L.M.; Bos, J.W.; Jansen, M.D.; Curial, C.A.; Silence, K.; et al. Anti-C2 Antibody ARGX-117 Inhibits Complement in a Disease Model for Multifocal Motor Neuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1107. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziadkowiak, E.; Nowakowska-Kotas, M.; Rałowska-Gmoch, W.; Budrewicz, S.; Koszewicz, M. Molecular, Electrophysiological, and Ultrasonographic Differences in Selected Immune-Mediated Neuropathies with Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 9180. https://doi.org/10.3390/ijms24119180
Dziadkowiak E, Nowakowska-Kotas M, Rałowska-Gmoch W, Budrewicz S, Koszewicz M. Molecular, Electrophysiological, and Ultrasonographic Differences in Selected Immune-Mediated Neuropathies with Therapeutic Implications. International Journal of Molecular Sciences. 2023; 24(11):9180. https://doi.org/10.3390/ijms24119180
Chicago/Turabian StyleDziadkowiak, Edyta, Marta Nowakowska-Kotas, Wiktoria Rałowska-Gmoch, Sławomir Budrewicz, and Magdalena Koszewicz. 2023. "Molecular, Electrophysiological, and Ultrasonographic Differences in Selected Immune-Mediated Neuropathies with Therapeutic Implications" International Journal of Molecular Sciences 24, no. 11: 9180. https://doi.org/10.3390/ijms24119180
APA StyleDziadkowiak, E., Nowakowska-Kotas, M., Rałowska-Gmoch, W., Budrewicz, S., & Koszewicz, M. (2023). Molecular, Electrophysiological, and Ultrasonographic Differences in Selected Immune-Mediated Neuropathies with Therapeutic Implications. International Journal of Molecular Sciences, 24(11), 9180. https://doi.org/10.3390/ijms24119180