
Genetically Engineered Mice Unveil In Vivo Roles of the Mediator Complex

 , ,
, ,
Abstract
:1. Introduction
2. Embryogenesis and Development
2.1. Preimplantation
2.2. Gastrulation
2.3. Neurulation and Organogenesis
2.4. After Birth
2.5. Contradictions
3. Metabolism
3.1. Liver Knockouts
3.2. Adipose Tissue Knockouts
3.3. Muscle Tissue Knockouts
4. Intestinal Knockout
5. Hematopoiesis and Immunity
6. Mediator Complex Subunits in Cardiomyocytes
7. Bone Homeostasis
8. Neurogenesis and Brain Function
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsai, K.-L.; Sato, S.; Tomomori-Sato, C.; Conaway, R.C.; Conaway, J.W.; Asturias, F.J. A Conserved Mediator–CDK8 Kinase Module Association Regulates Mediator–RNA Polymerase II Interaction. Nat. Struct. Mol. Biol. 2013, 20, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Freitas, K.A.; Belk, J.A.; Sotillo, E.; Quinn, P.J.; Ramello, M.C.; Malipatlolla, M.; Daniel, B.; Sandor, K.; Klysz, D.; Bjelajac, J.; et al. Enhanced T Cell Effector Activity by Targeting the Mediator Kinase Module. Science 2022, 378, eabn5647. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Nayak, S.; Iwasa, J.; Taatjes, D.J. The Mediator Complex as a Master Regulator of Transcription by RNA Polymerase II. Nat. Rev. Mol. Cell Biol. 2022, 23, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Hamidi, N.; Del Sol, R.; Benschop, J.J.; Nancy, T.; Li, C.; Francis, L.; Tzouros, M.; Krijgsveld, J.; Holstege, F.C.P.; et al. Suppression of Mediator Is Regulated by Cdk8-Dependent Grr1 Turnover of the Med3 Coactivator. Proc. Natl. Acad. Sci. USA 2014, 111, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 Is a Positive Regulator of Transcriptional Elongation within the Serum Response Network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef]
- Martinez-Fabregas, J.; Wang, L.; Pohler, E.; Cozzani, A.; Wilmes, S.; Kazemian, M.; Mitra, S.; Moraga, I. CDK8 Fine-Tunes IL-6 Transcriptional Activities by Limiting STAT3 Resident Time at the Gene Loci. Cell Rep. 2020, 33, 108545. [Google Scholar] [CrossRef]
- Li, L.; Walsh, R.M.; Wagh, V.; James, M.F.; Beauchamp, R.L.; Chang, Y.-S.; Gusella, J.F.; Hochedlinger, K.; Ramesh, V. Mediator Subunit Med28 Is Essential for Mouse Peri-Implantation Development and Pluripotency. PLoS ONE 2015, 10, e0140192. [Google Scholar] [CrossRef]
- Risley, M.D.; Clowes, C.; Yu, M.; Mitchell, K.; Hentges, K.E. The Mediator Complex Protein Med31 Is Required for Embryonic Growth and Cell Proliferation during Mammalian Development. Dev. Biol. 2010, 342, 146–156. [Google Scholar] [CrossRef]
- Li, N.; Fassl, A.; Chick, J.; Inuzuka, H.; Li, X.; Mansour, M.R.; Liu, L.; Wang, H.; King, B.; Shaik, S.; et al. Cyclin C Is a Haploinsufficient Tumor Suppressor. Nat. Cell Biol. 2014, 16, 1080–1091. [Google Scholar] [CrossRef]
- Willis, S.D.; Hanley, S.E.; Doyle, S.J.; Beluch, K.; Strich, R.; Cooper, K.F. Cyclin C-Cdk8 Kinase Phosphorylation of Rim15 Prevents the Aberrant Activation of Stress Response Genes. Front. Cell Dev. Biol. 2022, 10, 867257. [Google Scholar] [CrossRef]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dölken, L.; Strobl, B.; Müller, M.; Taatjes, D.J.; et al. CDK8 Kinase Phosphorylates Transcription Factor STAT1 to Selectively Regulate the Interferon Response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Vincent, O.; Kuchin, S.; Hong, S.-P.; Townley, R.; Vyas, V.K.; Carlson, M. Interaction of the Srb10 Kinase with Sip4, a Transcriptional Activator of Gluconeogenic Genes in Saccharomyces Cerevisiae. Mol. Cell. Biol. 2001, 21, 5790–5796. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B.; Győrffy, B.; Mack, Z.T.; Shtil, A.A.; Shtutman, M.S.; Chen, M.; Broude, E.V. Identifying Cancers Impacted by CDK8/19. Cells 2019, 8, 821. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.; Garabedian, M.J. The Mediator Complex in Genomic and Non-Genomic Signaling in Cancer. Steroids 2018, 133, 8–14. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Uehara, T.; Abe, K.; Oginuma, M.; Harako, S.; Ishitani, S.; Lehesjoki, A.-E.; Bierhals, T.; Kloth, K.; Ehmke, N.; et al. CDK19-Related Disorder Results from Both Loss-of-Function and Gain-of-Function de Novo Missense Variants. Genet. Med. 2021, 23, 1050–1057. [Google Scholar] [CrossRef]
- Spaeth, J.M.; Kim, N.H.; Boyer, T.G. Mediator and Human Disease. Semin. Cell Dev. Biol. 2011, 22, 776–787. [Google Scholar] [CrossRef]
- Caro-Llopis, A.; Rosello, M.; Orellana, C.; Oltra, S.; Monfort, S.; Mayo, S.; Martinez, F. De Novo Mutations in Genes of Mediator Complex Causing Syndromic Intellectual Disability: Mediatorpathy or Transcriptomopathy? Pediatr. Res. 2016, 80, 809–815. [Google Scholar] [CrossRef]
- Poot, M. Mutations in Mediator Complex Genes CDK8, MED12, MED13, and MEDL13 Mediate Overlapping Developmental Syndromes. Mol. Syndromol. 2019, 10, 239–242. [Google Scholar] [CrossRef]
- Uthe, H.; Vanselow, J.T.; Schlosser, A. Proteomic Analysis of the Mediator Complex Interactome in Saccharomyces Cerevisiae. Sci. Rep. 2017, 7, 43584. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator Kinase Inhibition Further Activates Super-Enhancer Associated Genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Balamotis, M.A.; Pennella, M.A.; Stevens, J.L.; Wasylyk, B.; Belmont, A.S.; Berk, A.J. Complexity in Transcription Control at the Activation Domain–Mediator Interface. Sci. Signal. 2009, 2, ra20. [Google Scholar] [CrossRef] [PubMed]
- Rocha, P.P.; Scholze, M.; Bleiss, W.; Schrewe, H. Med12 Is Essential for Early Mouse Development and for Canonical Wnt and Wnt/PCP Signaling. Dev. Camb. Engl. 2010, 137, 2723–2731. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Marcho, C.; Wang, Y.; Degani, R.; Golan, M.; Tremblay, K.D.; Rivera-Pérez, J.A.; Mager, J. Med20 Is Essential for Early Embryogenesis and Regulates Nanog Expression. Reprod. Camb. Engl. 2018, 157, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.; Waskow, C.; Krötschel, M.; van Essen, D.; Rodriguez, P.; Zhang, X.; Guyot, B.; Roeder, R.G.; Borggrefe, T. The Mediator Complex Functions as a Coactivator for GATA-1 in Erythropoiesis via Subunit Med1/TRAP220. Proc. Natl. Acad. Sci. USA 2006, 103, 18504–18509. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, Y.; Qi, Y.; Xu, Y.; Wang, S.; Chen, F.; Shen, M.; Chen, M.; Chen, N.; Yang, L.; et al. CDK19 Regulates the Proliferation of Hematopoietic Stem Cells and Acute Myeloid Leukemia Cells by Suppressing P53-Mediated Transcription of P21. Leukemia 2022, 36, 956–969. [Google Scholar] [CrossRef]
- Groza, T.; Gomez, F.L.; Mashhadi, H.H.; Muñoz-Fuentes, V.; Gunes, O.; Wilson, R.; Cacheiro, P.; Frost, A.; Keskivali-Bond, P.; Vardal, B.; et al. The International Mouse Phenotyping Consortium: Comprehensive Knockout Phenotyping Underpinning the Study of Human Disease. Nucleic Acids Res. 2023, 51, D1038–D1045. [Google Scholar] [CrossRef]
- Huszar, J.M.; Jia, Y.; Reddy, J.K.; Payne, C.J. Med1 Regulates Meiotic Progression during Spermatogenesis in Mice. Reprod. Camb. Engl. 2015, 149, 597–604. [Google Scholar] [CrossRef]
- Yoshizaki, K.; Hu, L.; Nguyen, T.; Sakai, K.; He, B.; Fong, C.; Yamada, Y.; Bikle, D.D.; Oda, Y. Ablation of Coactivator Med1 Switches the Cell Fate of Dental Epithelia to That Generating Hair. PLoS ONE 2014, 9, e99991. [Google Scholar] [CrossRef]
- Noguchi, F.; Nakajima, T.; Inui, S.; Reddy, J.K.; Itami, S. Alteration of Skin Wound Healing in Keratinocyte-Specific Mediator Complex Subunit 1 Null Mice. PLoS ONE 2014, 9, e102271. [Google Scholar] [CrossRef]
- Hasegawa, N.; Sumitomo, A.; Fujita, A.; Aritome, N.; Mizuta, S.; Matsui, K.; Ishino, R.; Inoue, K.; Urahama, N.; Nose, J.; et al. Mediator Subunits MED1 and MED24 Cooperatively Contribute to Pubertal Mammary Gland Development and Growth of Breast Carcinoma Cells. Mol. Cell. Biol. 2012, 32, 1483–1495. [Google Scholar] [CrossRef]
- Miao, Y.-L.; Gambini, A.; Zhang, Y.; Padilla-Banks, E.; Jefferson, W.N.; Bernhardt, M.L.; Huang, W.; Li, L.; Williams, C.J. Mediator Complex Component MED13 Regulates Zygotic Genome Activation and Is Required for Postimplantation Development in the Mouse. Biol. Reprod. 2018, 98, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Bhatt, S.; Sandell, L.L.; Seidel, C.W.; Ahn, Y.; Krumlauf, R.E.; Trainor, P.A. The Mediator Subunit, Med23 Is Required for Embryonic Survival and Regulation of Canonical WNT Signaling During Cranial Ganglia Development. Front. Physiol. 2020, 11, 531933. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Bhatt, S.; Falcon, K.T.; Sandell, L.L.; Trainor, P.A. Med23 Regulates Sox9 Expression during Craniofacial Development. J. Dent. Res. 2021, 100, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Westerling, T.; Kuuluvainen, E.; Mäkelä, T.P. Cdk8 Is Essential for Preimplantation Mouse Development. Mol. Cell. Biol. 2007, 27, 6177–6182. [Google Scholar] [CrossRef]
- Postlmayr, A.; Dumeau, C.E.; Wutz, A. Cdk8 Is Required for Establishment of H3K27me3 and Gene Repression by Xist and Mouse Development. Dev. Camb. Engl. 2020, 147, dev175141. [Google Scholar] [CrossRef]
- AOKI, F. Zygotic Gene Activation in Mice: Profile and Regulation. J. Reprod. Dev. 2022, 68, 79–84. [Google Scholar] [CrossRef]
- Huelsken, J.; Vogel, R.; Brinkmann, V.; Erdmann, B.; Birchmeier, C.; Birchmeier, W. Requirement for Beta-Catenin in Anterior-Posterior Axis Formation in Mice. J. Cell Biol. 2000, 148, 567–578. [Google Scholar] [CrossRef]
- Stumpf, M.; Yue, X.; Schmitz, S.; Luche, H.; Reddy, J.K.; Borggrefe, T. Specific Erythroid-Lineage Defect in Mice Conditionally Deficient for Mediator Subunit Med1. Proc. Natl. Acad. Sci. USA 2010, 107, 21541–21546. [Google Scholar] [CrossRef]
- Ito, M.; Okano, H.J.; Darnell, R.B.; Roeder, R.G. The TRAP100 Component of the TRAP/Mediator Complex Is Essential in Broad Transcriptional Events and Development. EMBO J. 2002, 21, 3464–3475. [Google Scholar] [CrossRef]
- Nakajima, T.; Inui, S.; Fushimi, T.; Noguchi, F.; Kitagawa, Y.; Reddy, J.K.; Itami, S. Roles of MED1 in Quiescence of Hair Follicle Stem Cells and Maintenance of Normal Hair Cycling. J. Investig. Dermatol. 2013, 133, 354–360. [Google Scholar] [CrossRef]
- Knuesel, M.T.; Meyer, K.D.; Donner, A.J.; Espinosa, J.M.; Taatjes, D.J. The Human CDK8 Subcomplex Is a Histone Kinase That Requires Med12 for Activity and Can Function Independently of Mediator. Mol. Cell. Biol. 2009, 29, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Luyties, O.; Taatjes, D.J. The Mediator Kinase Module: An Interface between Cell Signaling and Transcription. Trends Biochem. Sci. 2022, 47, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Orgilles, B.; Saldaña-Meyer, R.; Wang, E.; Trompouki, E.; Fassl, A.; Lau, S.; Mullenders, J.; Rocha, P.P.; Raviram, R.; Guillamot, M.; et al. MED12 Regulates HSC-Specific Enhancers Independently of Mediator Kinase Activity to Control Hematopoiesis. Cell Stem Cell 2016, 19, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Rachez, C.; Suldan, Z.; Ward, J.; Chang, C.P.; Burakov, D.; Erdjument-Bromage, H.; Tempst, P.; Freedman, L.P. A Novel Protein Complex That Interacts with the Vitamin D3 Receptor in a Ligand-Dependent Manner and Enhances VDR Transactivation in a Cell-Free System. Genes Dev. 1998, 12, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Fondell, J.D.; Ge, H.; Roeder, R.G. Ligand Induction of a Transcriptionally Active Thyroid Hormone Receptor Coactivator Complex. Proc. Natl. Acad. Sci. USA 1996, 93, 8329–8333. [Google Scholar] [CrossRef]
- Bai, L.; Jia, Y.; Viswakarma, N.; Huang, J.; Vluggens, A.; Wolins, N.E.; Jafari, N.; Rao, M.S.; Borensztajn, J.; Yang, G.; et al. Transcription Coactivator Mediator Subunit MED1 Is Required for the Development of Fatty Liver in the Mouse. Hepatology 2011, 53, 1164–1174. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, X.; Birsoy, K.; Roeder, R.G. A Muscle-Specific Knockout Implicates Nuclear Receptor Coactivator MED1 in the Regulation of Glucose and Energy Metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 10196–10201. [Google Scholar] [CrossRef]
- Ito, K.; Schneeberger, M.; Gerber, A.; Jishage, M.; Marchildon, F.; Maganti, A.V.; Cohen, P.; Friedman, J.M.; Roeder, R.G. Critical Roles of Transcriptional Coactivator MED1 in the Formation and Function of Mouse Adipose Tissues. Genes Dev. 2021, 35, 729–748. [Google Scholar] [CrossRef]
- Jia, Y.; Viswakarma, N.; Reddy, J.K. Med1 Subunit of the Mediator Complex in Nuclear Receptor-Regulated Energy Metabolism, Liver Regeneration, and Hepatocarcinogenesis. Gene Expr. 2014, 16, 63–75. [Google Scholar] [CrossRef]
- Amoasii, L.; Holland, W.; Sanchez-Ortiz, E.; Baskin, K.K.; Pearson, M.; Burgess, S.C.; Nelson, B.R.; Bassel-Duby, R.; Olson, E.N. A MED13-Dependent Skeletal Muscle Gene Program Controls Systemic Glucose Homeostasis and Hepatic Metabolism. Genes Dev. 2016, 30, 434–446. [Google Scholar] [CrossRef]
- Youn, D.Y.; Xiaoli, A.M.; Zong, H.; Okada, J.; Liu, L.; Pessin, J.; Pessin, J.E.; Yang, F. The Mediator Complex Kinase Module Is Necessary for Fructose Regulation of Liver Glycogen Levels through Induction of Glucose-6-Phosphatase Catalytic Subunit (G6pc). Mol. Metab. 2021, 48, 101227. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; van Rooij, E.; Johnson, B.A.; DeLeon, S.M.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A Cardiac MicroRNA Governs Systemic Energy Homeostasis by Regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Scotti, E.; Stoffel, M. CDK8 Regulates Insulin Secretion and Mediates Postnatal and Stress-Induced Expression of Neuropeptides in Pancreatic β Cells. Cell Rep. 2019, 28, 2892–2904.e7. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xiaoli, A.M.; Li, Y.; Siqin, G.; Wu, T.; Strich, R.; Pessin, J.E.; Yang, F. The Conserved Mediator Subunit Cyclin C (CCNC) Is Required for Brown Adipocyte Development and Lipid Accumulation. Mol. Metab. 2022, 64, 101548. [Google Scholar] [CrossRef]
- Youn, D.Y.; Xiaoli, A.M.; Kwon, H.; Yang, F.; Pessin, J.E. The Subunit Assembly State of the Mediator Complex Is Nutrient-Regulated and Is Dysregulated in a Genetic Model of Insulin Resistance and Obesity. J. Biol. Chem. 2019, 294, 9076–9083. [Google Scholar] [CrossRef]
- Dean, J.M.; He, A.; Tan, M.; Wang, J.; Lu, D.; Razani, B.; Lodhi, I.J. MED19 Regulates Adipogenesis and Maintenance of White Adipose Tissue Mass by Mediating PPARγ-Dependent Gene Expression. Cell Rep. 2020, 33, 108228. [Google Scholar] [CrossRef]
- Tang, W.-S.; Weng, L.; Wang, X.; Liu, C.-Q.; Hu, G.-S.; Yin, S.-T.; Tao, Y.; Hong, N.-N.; Guo, H.; Liu, W.; et al. The Mediator Subunit MED20 Organizes the Early Adipogenic Complex to Promote Development of Adipose Tissues and Diet-Induced Obesity. Cell Rep. 2021, 36, 109314. [Google Scholar] [CrossRef]
- Chu, Y.; Rosso, L.G.; Huang, P.; Wang, Z.; Xu, Y.; Yao, X.; Bao, M.; Yan, J.; Song, H.; Wang, G. Liver Med23 Ablation Improves Glucose and Lipid Metabolism through Modulating FOXO1 Activity. Cell Res. 2014, 24, 1250–1265. [Google Scholar] [CrossRef]
- Zhou, J.; Singh, B.K.; Ho, J.P.; Lim, A.; Bruinstroop, E.; Ohba, K.; Sinha, R.A.; Yen, P.M. MED1 Mediator Subunit Is a Key Regulator of Hepatic Autophagy and Lipid Metabolism. Autophagy 2021, 17, 4043–4061. [Google Scholar] [CrossRef]
- Jia, Y.; Qi, C.; Kashireddi, P.; Surapureddi, S.; Zhu, Y.-J.; Rao, M.S.; Le Roith, D.; Chambon, P.; Gonzalez, F.J.; Reddy, J.K. Transcription Coactivator PBP, the Peroxisome Proliferator-Activated Receptor (PPAR)-Binding Protein, Is Required for PPARα-Regulated Gene Expression in Liver. J. Biol. Chem. 2004, 279, 24427–24434. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, D.; Li, C.; Min, L.; Wang, G. Mediator MED23 Regulates Inflammatory Responses and Liver Fibrosis. PLoS Biol. 2019, 17, e3000563. [Google Scholar] [CrossRef] [PubMed]
- Daubas, P.; Tajbakhsh, S.; Hadchouel, J.; Primig, M.; Buckingham, M. Myf5 Is a Novel Early Axonal Marker in the Mouse Brain and Is Subjected to Post-Transcriptional Regulation in Neurons. Dev. Camb. Engl. 2000, 127, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Park, Y.-K.; Lee, J.-E.; Tran, N.; Gavrilova, O.; Ge, K. MED1 Is a Lipogenesis Coactivator Required for Postnatal Adipose Expansion. Genes Dev. 2021, 35, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liang, Y.; Park, J.Y.; Chen, D.; Yao, X.; Xiao, Q.; Liu, Z.; Jiang, B.; Fu, Y.; Bao, M.; et al. Mediator MED23 Plays Opposing Roles in Directing Smooth Muscle Cell and Adipocyte Differentiation. Genes Dev. 2012, 26, 2192–2205. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, L.; Huang, Y.; Yin, J.; Berk, A.J.; Friedman, J.M.; Wang, G. Mediator MED23 Links Insulin Signaling to the Adipogenesis Transcription Cascade. Dev. Cell 2009, 16, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yin, J.; Liang, Y.; Li, C.; Gao, P.; Yu, Y.; Wang, G. Mediator Med23 Deficiency in Smooth Muscle Cells Prevents Neointima Formation after Arterial Injury. Cell Discov. 2021, 7, 59. [Google Scholar] [CrossRef]
- Carrer, M.; Liu, N.; Grueter, C.E.; Williams, A.H.; Frisard, M.I.; Hulver, M.W.; Bassel-Duby, R.; Olson, E.N. Control of Mitochondrial Metabolism and Systemic Energy Homeostasis by MicroRNAs 378 and 378. Proc. Natl. Acad. Sci. USA 2012, 109, 15330–15335. [Google Scholar] [CrossRef]
- McCleland, M.L.; Soukup, T.M.; Liu, S.D.; Esensten, J.H.; de Sousa e Melo, F.; Yaylaoglu, M.; Warming, S.; Roose-Girma, M.; Firestein, R. Cdk8 Deletion in the Apc(Min) Murine Tumour Model Represses EZH2 Activity and Accelerates Tumourigenesis. J. Pathol. 2015, 237, 508–519. [Google Scholar] [CrossRef]
- Prieto, S.; Dubra, G.; Camasses, A.; Aznar, A.B.; Begon-Pescia, C.; Simboeck, E.; Pirot, N.; Gerbe, F.; Angevin, L.; Jay, P.; et al. CDK8 and CDK19 Act Redundantly to Control the CFTR Pathway in the Intestinal Epithelium. EMBO Rep. 2023, 24, e54261. [Google Scholar] [CrossRef]
- Dannappel, M.V.; Zhu, D.; Sun, X.; Chua, H.K.; Poppelaars, M.; Suehiro, M.; Khadka, S.; Sian, T.C.C.L.K.; Sooraj, D.; Loi, M.; et al. CDK8 and CDK19 Regulate Intestinal Differentiation and Homeostasis via the Chromatin Remodeling Complex SWI/SNF. J. Clin. Investig. 2022, 132, e158593. [Google Scholar] [CrossRef]
- Bai, L.; Li, Z.; Li, Q.; Guan, H.; Zhao, S.; Liu, R.; Wang, R.; Zhang, J.; Jia, Y.; Fan, J.; et al. Mediator 1 Is Atherosclerosis Protective by Regulating Macrophage Polarization. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Yang, X.; Su, Y.; Zheng, H.; Liu, J.; Liu, H.; Zou, Y.; Jiao, A.; Wang, X.; Zhang, C.; et al. Med1 Controls CD8 T Cell Maintenance through IL-7R-mediated Cell Survival Signalling. J. Cell. Mol. Med. 2021, 25, 4870–4876. [Google Scholar] [CrossRef] [PubMed]
- Jiao, A.; Liu, H.; Ding, R.; Zheng, H.; Zhang, C.; Feng, Z.; Lei, L.; Wang, X.; Su, Y.; Yang, X.; et al. Med1 Controls Effector CD8+ T Cell Differentiation and Survival through C/EBPβ-Mediated Transcriptional Control of T-Bet. J. Immunol. 2022, 209, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Fukuyama, T.; Brindle, P.K. T-Cells Null for the MED23 Subunit of Mediator Express Decreased Levels of KLF2 and Inefficiently Populate the Peripheral Lymphoid Organs. PLoS ONE 2014, 9, e102076. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, X.; Chen, X.; Liu, H.; Xu, Y.; Chu, Y.; Wang, G.; Liu, X. The Mediator Subunit Med23 Contributes to Controlling T-Cell Activation and Prevents Autoimmunity. Nat. Commun. 2014, 5, 5225. [Google Scholar] [CrossRef]
- Witalisz-Siepracka, A.; Gotthardt, D.; Prchal-Murphy, M.; Didara, Z.; Menzl, I.; Prinz, D.; Edlinger, L.; Putz, E.M.; Sexl, V. NK Cell-Specific CDK8 Deletion Enhances Antitumor Responses. Cancer Immunol. Res. 2018, 6, 458–466. [Google Scholar] [CrossRef]
- Grueter, C.E. Mediator Complex Dependent Regulation of Cardiac Development and Disease. Genom. Proteom. Bioinform. 2013, 11, 151–157. [Google Scholar] [CrossRef]
- Hall, D.D.; Spitler, K.M.; Grueter, C.E. Disruption of Cardiac Med1 Inhibits RNA Polymerase II Promoter Occupancy and Promotes Chromatin Remodeling. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H314–H325. [Google Scholar] [CrossRef]
- Tan, C.; Zhu, S.; Chen, Z.; Liu, C.; Li, Y.E.; Zhu, M.; Zhang, Z.; Zhang, Z.; Zhang, L.; Gu, Y.; et al. Mediator Complex Proximal Tail Subunit MED30 Is Critical for Mediator Core Stability and Cardiomyocyte Transcriptional Network. PLoS Genet. 2021, 17, e1009785. [Google Scholar] [CrossRef]
- Ponce, J.M.; Coen, G.; Spitler, K.M.; Dragisic, N.; Martins, I.; Hinton, A.; Mungai, M.; Tadinada, S.M.; Zhang, H.; Oudit, G.Y.; et al. Stress-Induced Cyclin C Translocation Regulates Cardiac Mitochondrial Dynamics. J. Am. Heart Assoc. 2020, 9, e014366. [Google Scholar] [CrossRef]
- Baskin, K.K.; Makarewich, C.A.; DeLeon, S.M.; Ye, W.; Chen, B.; Beetz, N.; Schrewe, H.; Bassel-Duby, R.; Olson, E.N. MED12 Regulates a Transcriptional Network of Calcium-Handling Genes in the Heart. JCI Insight 2017, 2, e91920. [Google Scholar] [CrossRef] [PubMed]
- Krebs, P.; Fan, W.; Chen, Y.-H.; Tobita, K.; Downes, M.R.; Wood, M.R.; Sun, L.; Li, X.; Xia, Y.; Ding, N.; et al. Lethal Mitochondrial Cardiomyopathy in a Hypomorphic Med30 Mouse Mutant Is Ameliorated by Ketogenic Diet. Proc. Natl. Acad. Sci. USA 2011, 108, 19678–19682. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Fukasawa, K.; Horie, T.; Kadota, T.; Lyu, J.; Tokumura, K.; Ochiai, S.; Iwahashi, S.; Suzuki, A.; Park, G.; et al. The Role of CDK8 in Mesenchymal Stem Cells in Controlling Osteoclastogenesis and Bone Homeostasis. Stem Cell Rep. 2022, 17, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, G.; Schiller, P.C.; Ricordi, C.; Roos, B.A.; Howard, G.A. Age-Related Osteogenic Potential of Mesenchymal Stromal Stem Cells from Human Vertebral Bone Marrow. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1999, 14, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Roles of Runx2 in Skeletal Development. Adv. Exp. Med. Biol. 2017, 962, 83–93. [Google Scholar] [CrossRef]
- Liu, Z.; Yao, X.; Yan, G.; Xu, Y.; Yan, J.; Zou, W.; Wang, G. Mediator MED23 Cooperates with RUNX2 to Drive Osteoblast Differentiation and Bone Development. Nat. Commun. 2016, 7, 11149. [Google Scholar] [CrossRef]
- Uehara, T.; Abe, K.; Oginuma, M.; Ishitani, S.; Yoshihashi, H.; Okamoto, N.; Takenouchi, T.; Kosaki, K.; Ishitani, T. Pathogenesis of CDK8-Associated Disorder: Two Patients with Novel CDK8 Variants and in Vitro and in Vivo Functional Analyses of the Variants. Sci. Rep. 2020, 10, 17575. [Google Scholar] [CrossRef]
- Kaufmann, R.; Straussberg, R.; Mandel, H.; Fattal-Valevski, A.; Ben-Zeev, B.; Naamati, A.; Shaag, A.; Zenvirt, S.; Konen, O.; Mimouni-Bloch, A.; et al. Infantile Cerebral and Cerebellar Atrophy Is Associated with a Mutation in the MED17 Subunit of the Transcription Preinitiation Mediator Complex. Am. J. Hum. Genet. 2010, 87, 667–670. [Google Scholar] [CrossRef]
- Clark, R.D.; Graham, J.M.; Friez, M.J.; Hoo, J.J.; Jones, K.L.; McKeown, C.; Moeschler, J.B.; Raymond, F.L.; Rogers, R.C.; Schwartz, C.E.; et al. FG Syndrome, an X-Linked Multiple Congenital Anomaly Syndrome: The Clinical Phenotype and an Algorithm for Diagnostic Testing. Genet. Med. 2009, 11, 769–775. [Google Scholar] [CrossRef]
- Snijders Blok, L.; Hiatt, S.M.; Bowling, K.M.; Prokop, J.W.; Engel, K.L.; Cochran, J.N.; Bebin, E.M.; Bijlsma, E.K.; Ruivenkamp, C.A.L.; Terhal, P.; et al. De Novo Mutations in MED13, a Component of the Mediator Complex, Are Associated with a Novel Neurodevelopmental Disorder. Hum. Genet. 2018, 137, 375–388. [Google Scholar] [CrossRef]
- Hashimoto, S.; Boissel, S.; Zarhrate, M.; Rio, M.; Munnich, A.; Egly, J.-M.; Colleaux, L. MED23 Mutation Links Intellectual Disability to Dysregulation of Immediate Early Gene Expression. Science 2011, 333, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- Trehan, A.; Brady, J.M.; Maduro, V.; Bone, W.P.; Huang, Y.; Golas, G.A.; Kane, M.S.; Lee, P.R.; Thurm, A.; Gropman, A.L.; et al. MED23-Associated Intellectual Disability in a Non-Consanguineous Family. Am. J. Med. Genet. A 2015, 167, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Y.; Zhang, S.; Li, C.-H.; Qi, C.-C.; Wang, Y.-Z.; Chen, J.-Y.; Wang, G.; Ding, Y.-Q.; Su, C.-J. Mediator Med23 Regulates Adult Hippocampal Neurogenesis. Front. Cell Dev. Biol. 2020, 8, 699. [Google Scholar] [CrossRef] [PubMed]
- Rubinato, E.; Rondeau, S.; Giuliano, F.; Kossorotoff, M.; Parodi, M.; Gherbi, S.; Steffan, J.; Jonard, L.; Marlin, S. MED12 Missense Mutation in a Three-Generation Family. Clinical Characterization of MED12-Related Disorders and Literature Review. Eur. J. Med. Genet. 2020, 63, 103768. [Google Scholar] [CrossRef] [PubMed]
- Vogl, M.R.; Reiprich, S.; Küspert, M.; Kosian, T.; Schrewe, H.; Nave, K.-A.; Wegner, M. Sox10 Cooperates with the Mediator Subunit 12 during Terminal Differentiation of Myelinating Glia. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 6679–6690. [Google Scholar] [CrossRef]
- Huang, T.-W.; Iyer, A.A.; Manalo, J.M.; Woo, J.; Bosquez Huerta, N.A.; McGovern, M.M.; Schrewe, H.; Pereira, F.A.; Groves, A.K.; Ohlemiller, K.K.; et al. Glial-Specific Deletion of Med12 Results in Rapid Hearing Loss via Degradation of the Stria Vascularis. J. Neurosci. 2021, 41, 7171–7181. [Google Scholar] [CrossRef]
- Clarke, P.A.; Ortiz-Ruiz, M.-J.; TePoele, R.; Adeniji-Popoola, O.; Box, G.; Court, W.; Czasch, S.; El Bawab, S.; Esdar, C.; Ewan, K.; et al. Assessing the Mechanism and Therapeutic Potential of Modulators of the Human Mediator Complex-Associated Protein Kinases. eLife 2016, 5, e20722. [Google Scholar] [CrossRef]
- Chen, M.; Li, J.; Liang, J.; Thompson, Z.S.; Kathrein, K.; Broude, E.V.; Roninson, I.B. Systemic Toxicity Reported for CDK8/19 Inhibitors CCT251921 and MSC2530818 Is Not Due to Target Inhibition. Cells 2019, 8, 1413. [Google Scholar] [CrossRef]
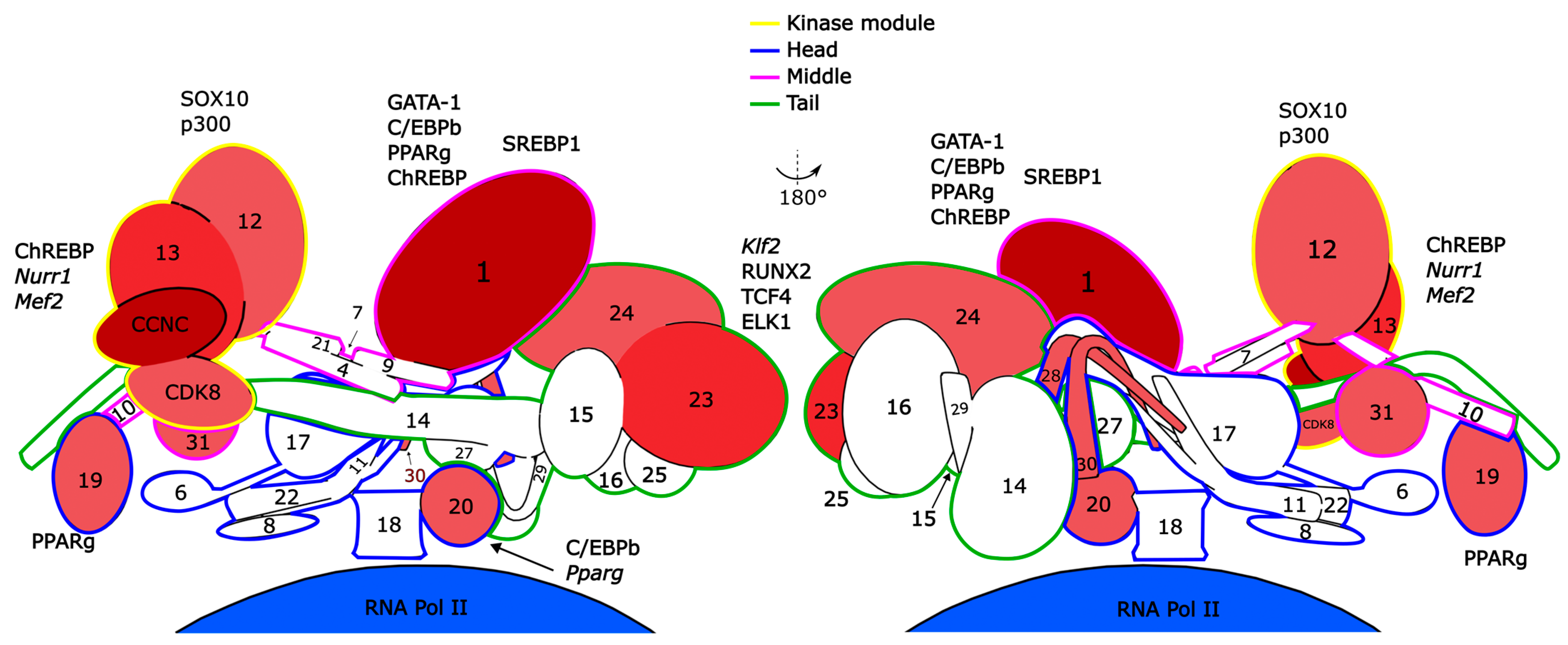
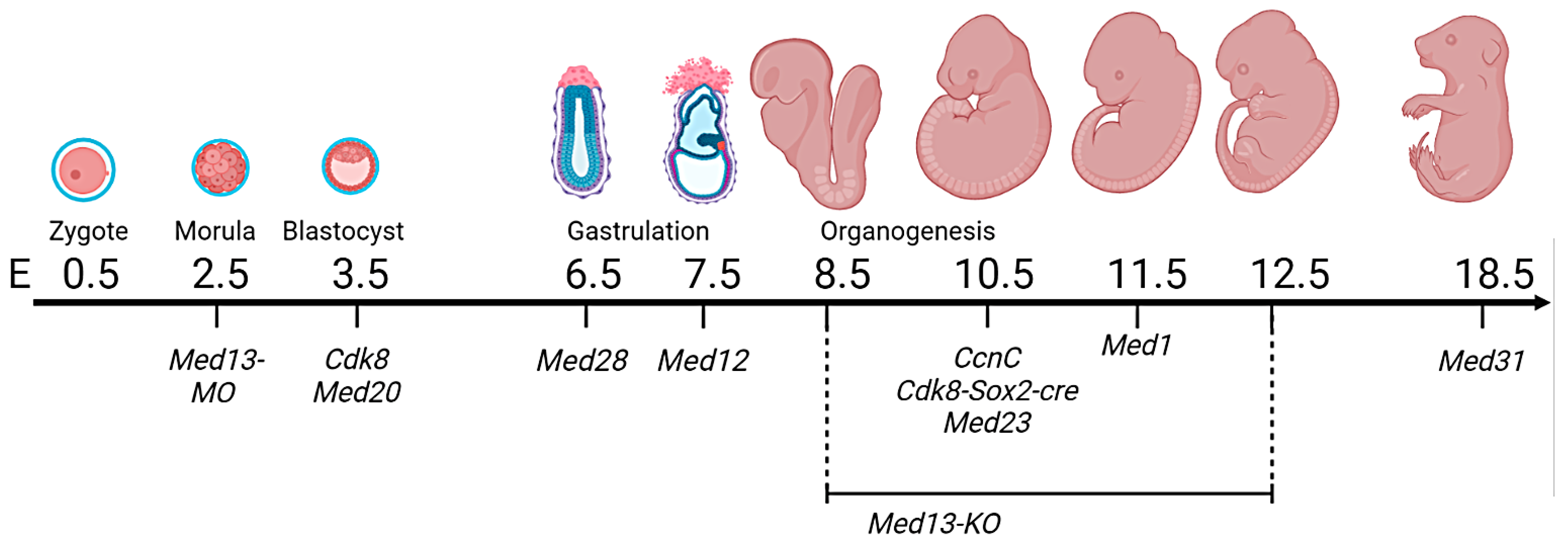


{kind=link}
{kind=link}
{kind=link}
| Protein | Type of Gene Inactivation | Death | Details | Reference |
|---|---|---|---|---|
| MED 1 | Vasa-Cre embryonic germ cells | Male mice with Med1 KO in germ cells were fertile; however, accelerated dynamics of prophase I of meiosis was observed: germ cells prematurely appeared in zygonema and pachynema stages | Huszar, 2015 [27] | |
| K14-Cre ectoderm and its derivatives | Formation of “hairy” teeth due to the transformation of dental epithelium into the epidermal epithelium. Ablation of Med1 resulted in suppression of Notch1 signaling. Suppression of Notch1 resulted in maintaining the cells in undifferentiated state, and Sox2 expression. Calcium action on the less-differentiated cells resulted in their differentiation into the hair epidermis | Yoshizaki, 2014 [28] | ||
| K5-Cre, epithelium | Young mice showed significantly accelerated wound closure and healing. The paracrine action of activin A was augmented due to suppressed follistatin expression, which induced phosphorylation of ERK and JNK and activation of the MAPK pathway | Noguchi, 2014 [29] | ||
| Constitutive | E11.5 1 | Med1−/− embryos at E10.5 were anemic; however, they had normal numbers of hematopoietic progenitor cells. Defects in forming erythroid burst-forming units and colony-forming units in these embryos were observed. MED1 interacted with the erythroid master regulator GATA-1. MED1 deficiency led to a defect in GATA-1-mediated transactivation | Stumpf, 2006 [24] | |
| MED1/MED24 | Constitutive, double heterozygous | Pubertal mammary glands showed significant developmental retardation. The Mediator partially failed to transduce signal from estrogen receptor bound to promoters to its target. The effect was not observed in pubertal uteri, nor in gestating glands | Hasegawa, 2012 [30] | |
| MED12 | Constitutive | E7.5 | Impaired mesoderm formation. Many embryos were arrested at pre-streak stages. Wnt/β-catenin signaling was abrogated | Rocha, 2010 [22] |
| Hypomorphic | E9.5 | Various morphological defects Wnt/β-catenin signaling pathway was abrogated | Rocha, 2010 [22] | |
| MED13 | Med13-MO knockdown by morpholino injection at the zygote stage | E2.5 | Delayed embryo development. Potentially, MED13 affected Zygotic genome activation, DNA reparation and mitosis progression | Miao, 2018 [31] |
| Constitutive | Before E12.5 | All pregnant females carrying Med13Δ/Δ embryos lost weight between E8.5 and E12.5. No Med13-KO were found at E12.5 | ||
| MED 20 | Constitutive | E3.5 | The epiblast marker Nanog was ectopically expressed in the trophectoderm of Med20 mutants, indicative of defects in trophoblast specification. Impaired implantation | Cui, 2018 [23] |
| MED23 | Constitutive | E9–10.5 | Mutant embryos died between embryonic E9.5 and E10.5, all three germ layers developed, and early organogenesis was initiated before death, which was likely caused by systemic circulatory failure. Med23 KO nearly eliminated Egr1 transcription in embryonic stem (ES) cells, leaving a paused polymerase at the promoter | Balamotis, 2009 [21] |
| Med23sn/sn (snouty mutation, single base pair nonsynonymous mutation in Exon 22 | E10.5 | Craniofacial abnormalities, including malformed frontonasal prominences, pharyngeal arches, otic and optic vesicles were observed. WNT/β-catenin signaling | Dash, 2020 [32] | |
| Med23bgeo/bgeo (genetrap between exons 12 and 13) | E10.5 | Developmental delay, failure to undergo axial turning | Dash, 2020 [32] | |
| Wnt1-Cre, neural crest cells | P0 | Micrognathia, glossoptosis, and cleft palate were observed. Sox9 mRNA and protein levels were both upregulated in neural crest cell-derived mesenchyme surrounding Meckel’s cartilage and in the palatal shelves | Dash, 2021 [33] | |
| Tek-Cre, vascular endothelium | E16.5 | Hemorrhage and edema | Dash, 2020 [32] | |
| MED28 | Protamine-Cre | E6.5 | MED28-deficiency caused the loss of pluripotency of the inner cell mass accompanied by reduced expression of key pluripotency transcription factors Oct4 and Nanog | Li, 2015 [7] |
| MED31 | Constitutive truncation | Prenatal lethality, around E18.5 | Defective or delayed chondrogenesis due to a lack of Sox9 and Col2a1 expression. MED31 mutant embryos had fewer proliferating cells than controls. Delayed embryo development was observed after the E8.5 stage. | Risley, 2010 [8] |
| CDK8 | Constitutive genetrap insertion in intron 4 of Cdk8 | E3.5 | Blastomere fragmentation, disrupted morula compaction | Westerling, 2007 [34] |
| Sox2-Cre Epiblast | Around E10.5 | Delayed embryos development. CDK8 was potentially involved in the X-chromosome inactivation | Postlmayr, 2020 [35] | |
| CCNC | Constitutive | E10.5 | Severe developmental retardation of mutant embryos, underdeveloped placental labyrinth layer | Li, 2014 [9] |
| Gene | Activator (Name, Cell Type) | Phenotype | Mechanism (TFs) | Reference |
|---|---|---|---|---|
| Med1 | Albumin-Cre, liver | Liver steatosis protection. Glucose tolerance, insulin sensitivity | PPARγ functional impairment | Bai, 2011 [46] |
| MCK-Cre, muscle | Obesity resistance. Glucose tolerance, insulin sensitivity. Switch towards slow muscle fibers | C/EBPα, PPARγ; UCP-1 and Cidea↑ 1 | Chen, 2010 [47] | |
| Myf5-Cre, dermomyotome derivatives at E10.5, then brown adipocytes | Growth retardation, weaning age death. Significant BAT mass reduction at E17.5 | Pparγ impaired. C/ebpα, Adipoq. Ucp1, Dio2, and Cox8b↓ 1 | Ito, 2021 [48] | |
| Adipoq-Cre, WAT | Nearly complete WAT loss by 6 wk age, mild decrease in BAT. Hepatic steatosis, insulin resistance | -”- | ||
| Albumin-Cre, liver | Diminished cellular proliferation. Abrogation of peroxisome proliferative response | PPARα | Jia, 2004 [49] | |
| Med13 | Myo-Cre, skeletal muscle | Hepatic steatosis resistance due to improved glucose clearance (on HFD) and reduced insulin level | NURR1, MEF2, GLUT4 ↑ 1 | Amoasii, 2016 [50] |
| Albumin-Cre, liver | Unchanged insulin sensitivity. Glycogen accumulation defect when fed fructose | G6PC, ChREBP, FOXO1 | Youn, 2021 [51] | |
| αMHC-Cre, cardiomyocyte | More susceptible to diet-induced obesity, metabolic syndrome | SREBP, RXR, PPARγ Thrsp, Gpd2, Eno1, Aacs | Grueter, 2012 [52] | |
| Cdk8 | Rip-Cre, pancreatic β cells | Higher glucose clearance under normal diet. Increased β cell sensitivity. Ectopic expression of neuropeptides in the cells | OSBPL3 lack of phosphorylation by CDK8 leads to insulin secretion. KO allows neuropeptide secretion to rescue cells from apoptosis. | Xue, 2019 [53] |
| Ccnc | Myf5-Cre, dermomyotome derivatives at E10.5, then brown adipocytes | BAT paucity, neonatal lethality | C/EBPα, GLUT4, ChREBP | Song, 2022 [54] |
| Ucp1-Cre, BAT | Browning of WAT upon cold exposure. Less lipid accumulation in BAT on normal diet | Akt2 | -”- | |
| Adipoq-Cre, all adipose tissue | Less lipid accumulation in BAT on normal diet | -”- | ||
| AAV8-Tbg-Cre, liver | No phenotype described | mTORC1 pathway | Youn, 2019 [55] | |
| Med19 | Adiponectin-Cre, adipose tissue | Loss of WAT, whitening of brown fat, hepatic steatosis, and insulin resistance | PPARγ | Dean, 2020 [56] |
| Med20 | Pdgfra-Cre, preadipocytes | Neonatal death. Severe BAT atrophy. Heterozygous mice are born normally and resist obesity on HFD. Glucose tolerance | MED20 bridges C/EBPβ to Pol II to promote transcription of PPARγ | Tang, 2021 [57] |
| Med23 | Albumin-Cre, liver | Improved glucose and lipid metabolism, insulin sensitivity | FOXO1 target genes (Irs2, Igfbp1, Pck1, G6pc) | Chu, 2014 [58] |
| Gene | Activator Details, Cell Type | Phenotype | Molecular Mechanism (TF) | Study |
|---|---|---|---|---|
| Med1 | Mx-Cre, Splenocytes, bone marrow, inducible | Block of erythroid development | GATA-1, TFIIB, absence of β-globin gene expression | Stumpf, 2010 [38] |
| Lyz2-Cre; myeloid lineage, macrophages; ApoE−/− background | Atherosclerosis enhancement; Med1 overexpression protective | PPARγ↓ 1 H3K4me1 and H3K27ac↑ 1 at M2 marker genes | Bai, 2017 [71] | |
| CD8+ T cells | Expansion↓ Killer population↓ Apoptosis↑ | T-bet– and Zeb2 transcriptional programs↓ C/EBPβ | Jiao, 2022 [73] | |
| Lck-Cre T-cells | CD8+ T cell↓ in spleen | IL-7Rα↓ pSTAT5↓ BIM↑ | Lei, 2021 [72] | |
| Med23 | Lck-Cre T-cells | T-cells failed to efficiently populate the peripheral lymphoid organs | Egr1, Egr2, Cd52↓ KLF2↓ | Kasper, 2014 [74] |
| Cd4Cre T-cells | T-cell hyperactivation, autoimmune syndrome | Egr1, Egr2 and Klf2↓ | Sun, 2014 [75] | |
| Med12 | Vav-Cre blood cells and HSPC Mx-Cre Splenocytes, bone marrow, inducible CreER ubiquitous | Bone marrow aplasia | p300 and CBP occupancy of c-kit enhancer | Aranda-Orgiles, 2016 [43] |
| Cdk19 | Constitutive | Proliferation of hematopoietic stem cells under stress is impaired | P53 dependent expression of p21 | Zhang, 2022 [25] |
| Ccnc | Mx1-Cre Splenocytes, bone marrow, inducible | T-cell-acute lymphoblastic leukemia, oncogenic Increased differentiation in T-cell lineage | ICN1 phosphorylation↓ stability↑ | Li, 2014 [9] |
| Cdk8 | Ncr1-Cre, NK-cells | Increased cytotoxicity | Expression of cytotoxic genes↑ | Witalisz-Siepracka, 2018 [76] |
| Gene | Activator Details, Cell Type | Phenotype | Mechanism | Reference |
|---|---|---|---|---|
| Med1 | αMHC-Cre, cardiomyocyte | Fibrosis↑ 1, early lethality, cardiac contractility↓ 1 | Deficit in RNA polymerase II recruitment to gene promoters, chromatin accessibility↓ 1 | Hall, 2019 [78] |
| Med12 | cKO αMHC-Cre | Dilated cardiomyopathy, cardiac contractility↓ | Atp2a2, Gja1, Gja5, Kcnn1, Pln, Ryr2, and Tnnt1 ↓; Cacna1d, Casq1, Gja3, and Slc8a2 ↑—altered expression of calcium-handling genes | Baskin, 2017 [81] |
| Med30 | cKO cTNT-Cre, icKO αMHC-Cre | Early lethality, mitochondrial cardiomyopathy | Mediator core stability (MED 4, 8, 14, 16, 17, 18, 24, 29, 31)↓ Tnni2, Mhy7, Gja1, Gaj5, Pln, Mycn, and Hey2↓ | Tan, 2021 [79] |
| Med30 | I44F protein substitution | Heart failure, fibrosis, focal myocardial necrosis, myofibril loss, progressive and selective decline in the transcription of genes necessary for OXPHOS and mitochondrial integrity, eventually leading to cardiac failure | Disturbed interactions of the ERRα, PGC-1α and Mediator complex with target genes | Krebs, 2011 [82] |
| Ccnc | cKO αMHC-Cre background | Heart mass↑ cardiac contractility↓ | Elongated mitochondria | Ponce, 2020 [80] |
| Ccnc | Overexpression | Heart failure, heart hypertrophy | Mitochondrial fission ↑ | Ponce, 2020 [80] |
| Gene | Activator (Name, Cell Type) | Phenotype | Molecular Mechanism (TF) | Reference |
|---|---|---|---|---|
| Med23 | Nestin-Cre/ER Inducible KO in neural stem cells | Proliferation of neural stem cells ↑ 1, cell cycle shortening Neuroblasts and immature neurons number in dentate gyrus ↓1, defective dendritic morphogenesis, deficiency in spatial and contextual fear memory | Reduction in cell cycle length of NSCs with unchanged progenitors. Enrichment in genes involved in cell proliferation (Sat1, Tnf, Igf1, Cd37, and Ereg), early response genes (such as Egr1, Egr2, and Egr4) expression ↓ 1 | Chen, 2020 [93] |
| Med12 | Aldh1l1-Cre/ER, astrocytes | Rapid hearing loss, stria vascularis degeneration, disorganization of basal cells adjacent to the spiral ligament, stria vascularis, leading to endolymph ionic imbalance, hair cell function disorder and subsequent hearing loss | Reduced interaction between MED12 and SOX10 leads to incorrect differentiation of oligodendrocytes; Tjp1, Cdh1, and Gjb3 genes expression disturbances | Huang, 2021 [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilchuk, L.A.; Kubekina, M.V.; Okulova, Y.D.; Silaeva, Y.Y.; Tatarskiy, V.V.; Filatov, M.A.; Bruter, A.V. Genetically Engineered Mice Unveil In Vivo Roles of the Mediator Complex. Int. J. Mol. Sci. 2023, 24, 9330. https://doi.org/10.3390/ijms24119330
Ilchuk LA, Kubekina MV, Okulova YD, Silaeva YY, Tatarskiy VV, Filatov MA, Bruter AV. Genetically Engineered Mice Unveil In Vivo Roles of the Mediator Complex. International Journal of Molecular Sciences. 2023; 24(11):9330. https://doi.org/10.3390/ijms24119330
Chicago/Turabian StyleIlchuk, Leonid A., Marina V. Kubekina, Yulia D. Okulova, Yulia Yu. Silaeva, Victor V. Tatarskiy, Maxim A. Filatov, and Alexandra V. Bruter. 2023. "Genetically Engineered Mice Unveil In Vivo Roles of the Mediator Complex" International Journal of Molecular Sciences 24, no. 11: 9330. https://doi.org/10.3390/ijms24119330