
Epigenetic Landscape Is Largely Shaped by Diversiform Transposons in Aegilops tauschii

Abstract
:1. Introduction
2. Results
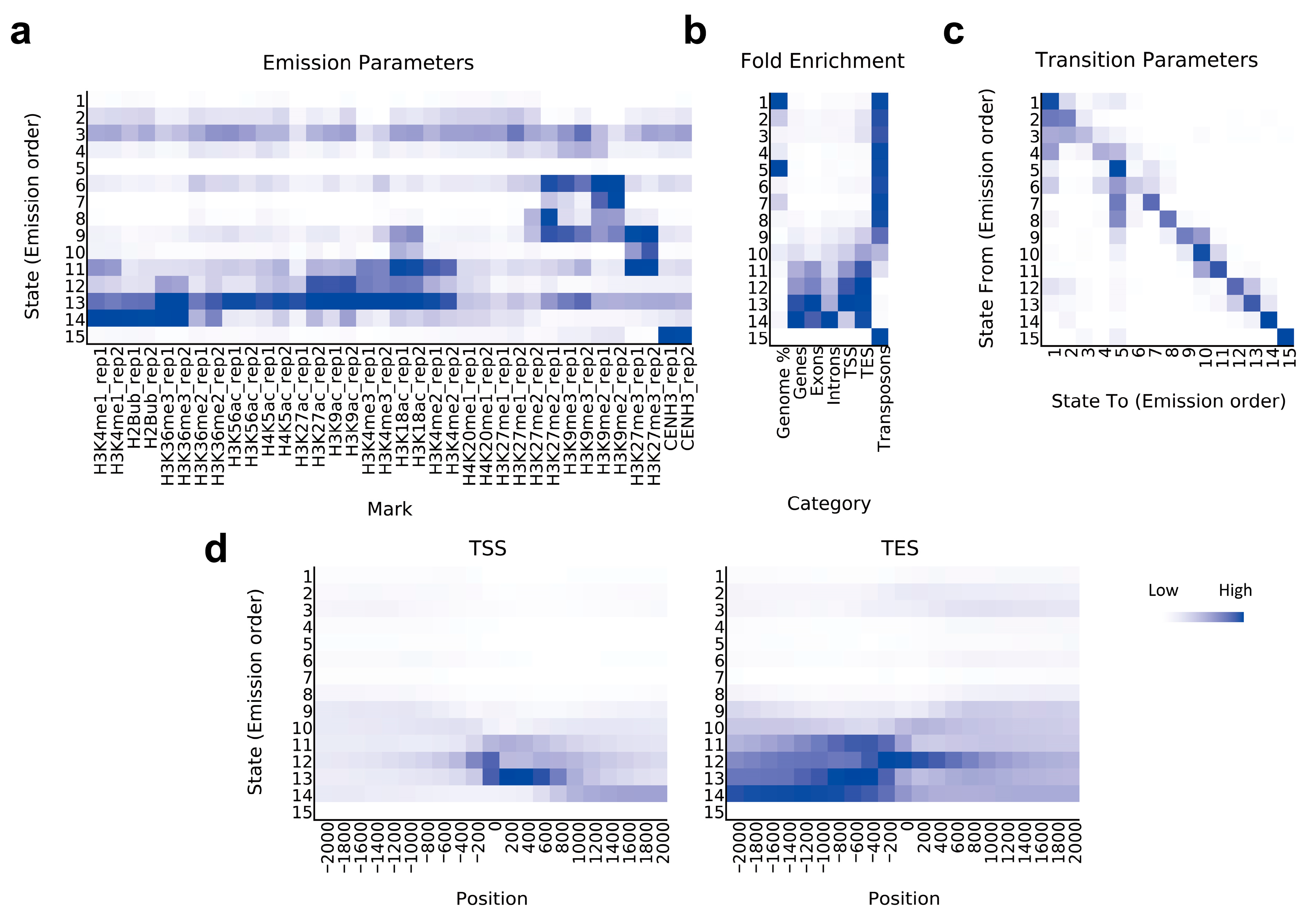
2.1. Chromatin State Profiling of Ae. tauschii
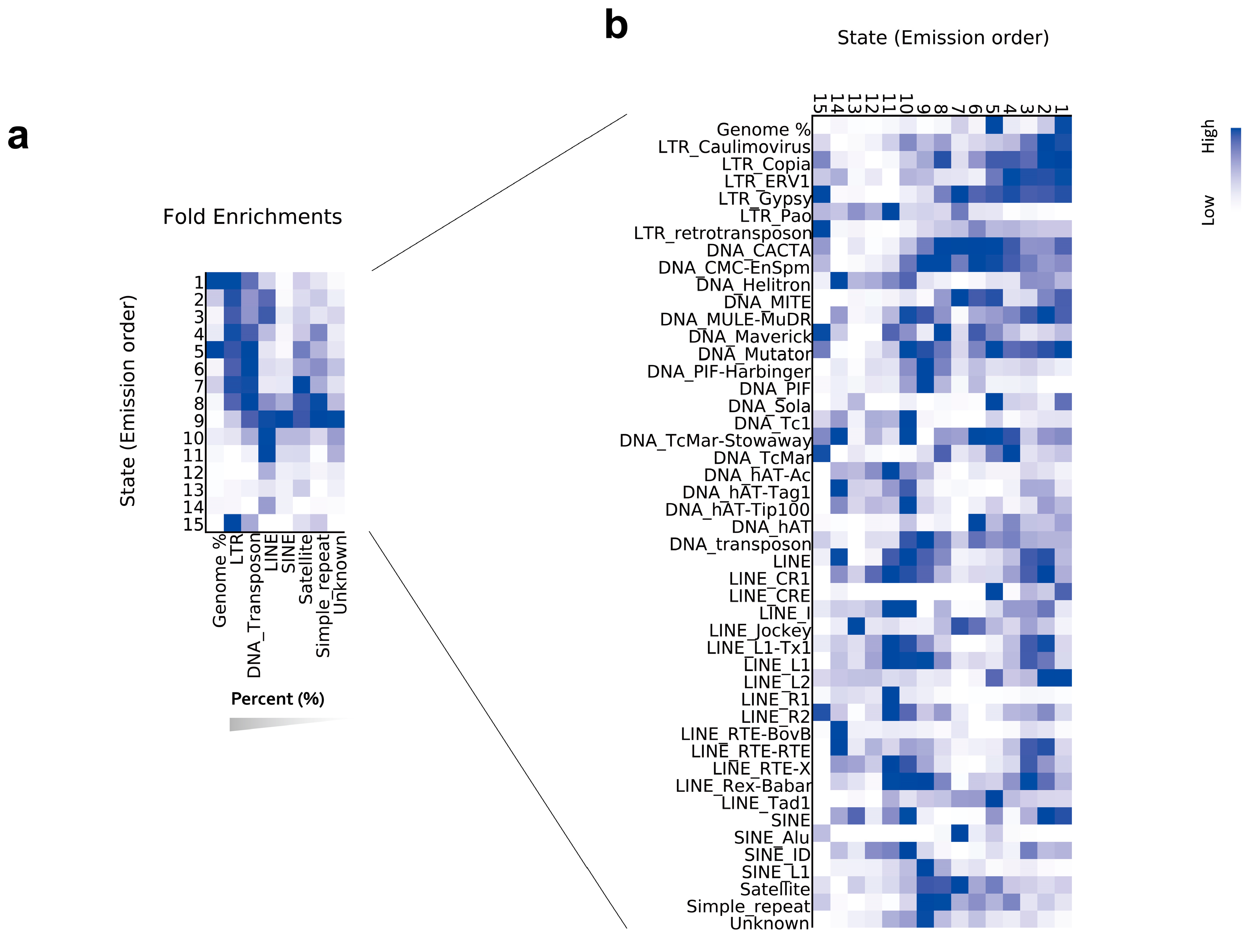
2.2. Chromatin State Signatures on TE Orders
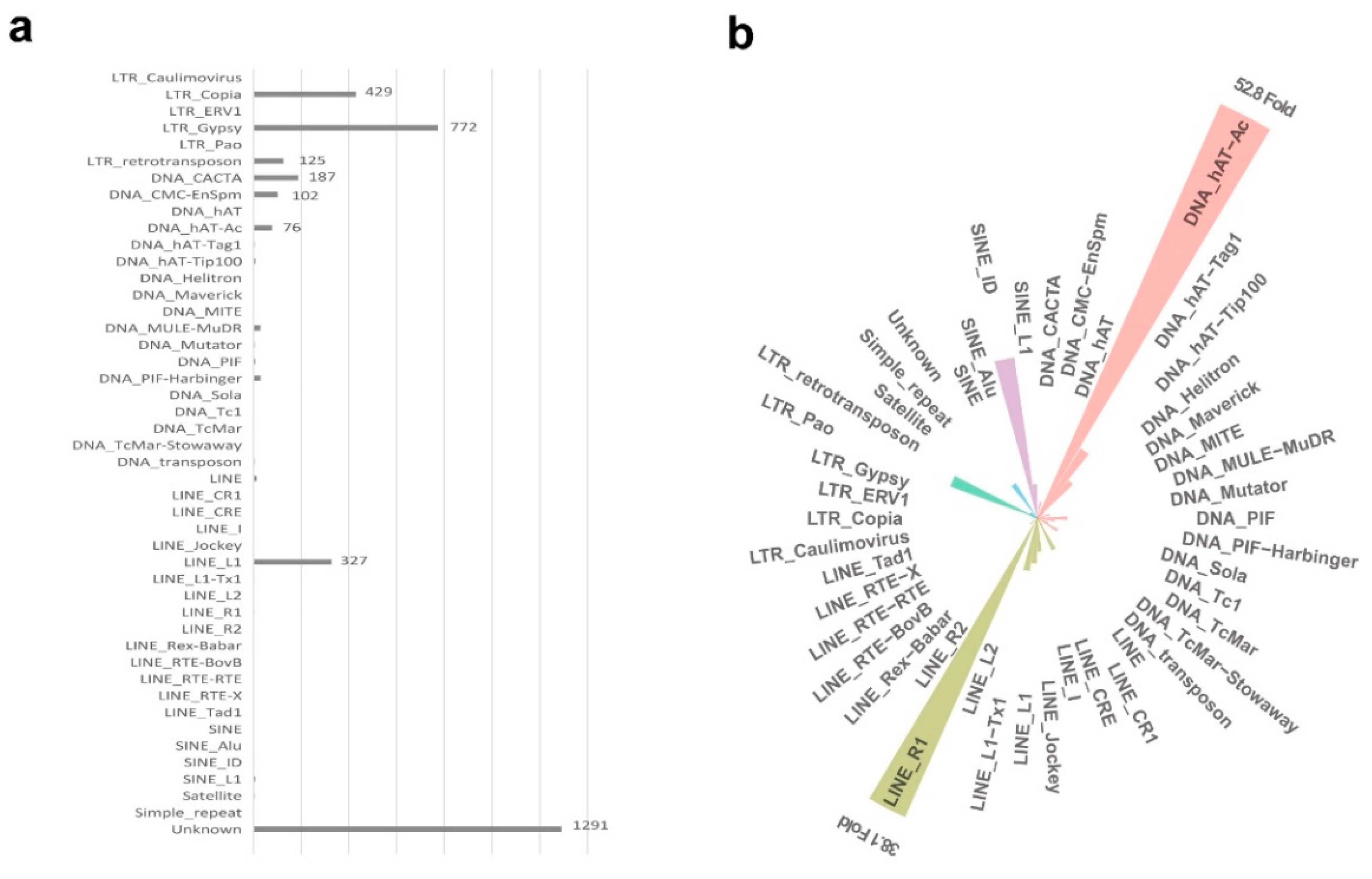
2.3. Chromatin State Signatures on TE Superfamilies
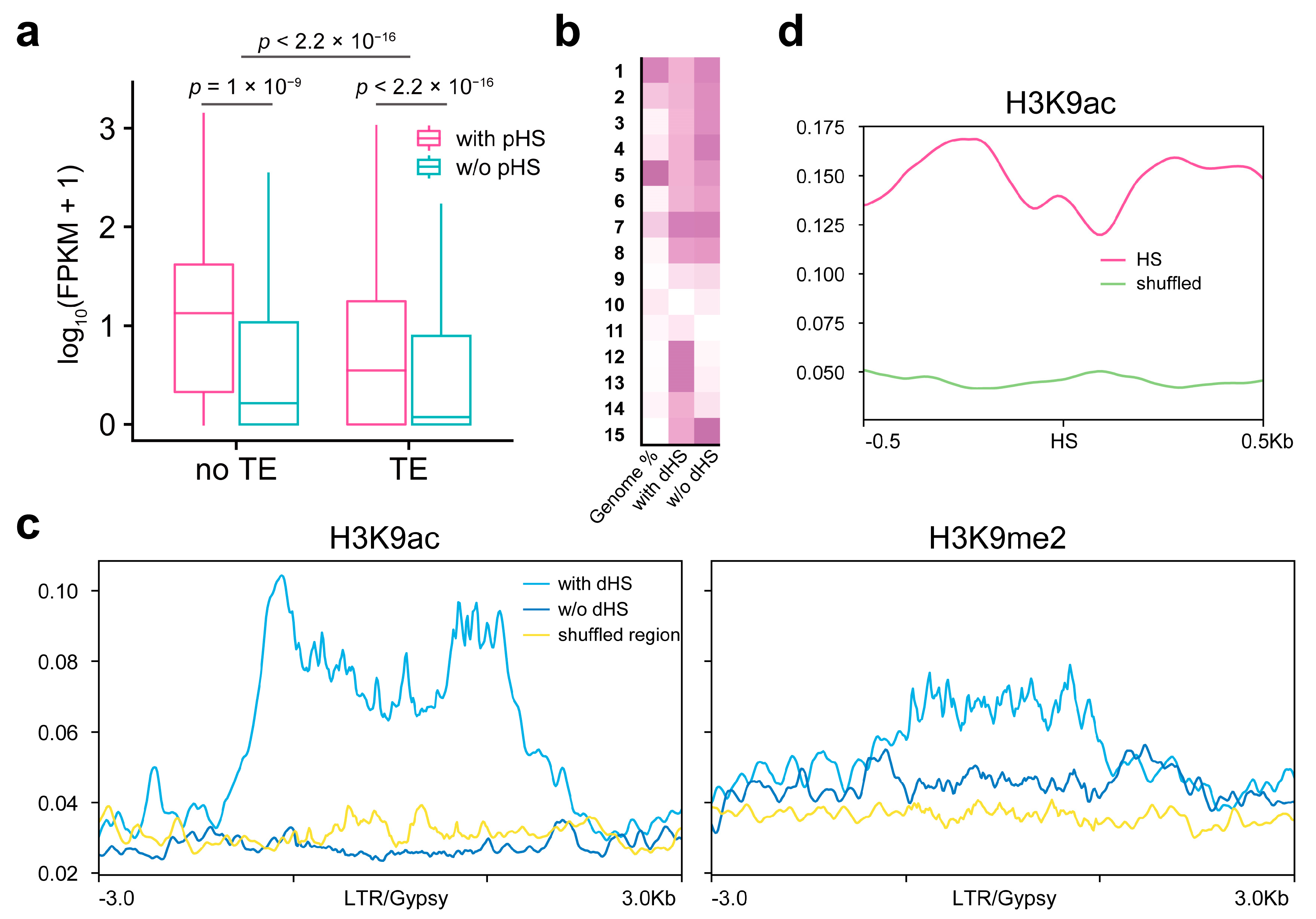
2.4. Chromatin Accessibility and Transcription of Chromatin States
2.5. Open Chromatin Regions on TEs

2.6. Impacts of TE Chromatin States on Openness and Gene Expression
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. ChIP-Seq and MNase-Seq
4.3. Learning of Chromatin States in Ae. tauschii
4.4. Identification and Classification of Open Chromatin Regions
4.5. Identification of Chromatin States on TE Orders and Superfamilies
4.6. Calculation of State and HS Enrichments on TEs
4.7. Prediction and Similarity Quantitation of Motifs
4.8. Identification of TE-Associated Chromatin States in Arabidopsis, Rice, and Maize
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Sousa, T.; Ribeiro, M.; Sabenca, C.; Igrejas, G. The 10,000-Year Success Story of Wheat! Foods 2021, 10, 2124. [Google Scholar] [CrossRef] [PubMed]
- Dubcovsky, J.; Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 2007, 316, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yao, Y.; Xin, M.; Peng, H.; Ni, Z.; Sun, Q. Shaping polyploid wheat for success: Origins, domestication, and the genetic improvement of agronomic traits. J. Integr. Plant Biol. 2022, 64, 536–563. [Google Scholar] [CrossRef]
- International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Pont, C.; Leroy, T.; Seidel, M.; Tondelli, A.; Duchemin, W.; Armisen, D.; Lang, D.; Bustos-Korts, D.; Goue, N.; Balfourier, F.; et al. Tracing the ancestry of modern bread wheats. Nat. Genet. 2019, 51, 905–911. [Google Scholar] [CrossRef]
- Zhao, G.; Zou, C.; Li, K.; Wang, K.; Li, T.; Gao, L.; Zhang, X.; Wang, H.; Yang, Z.; Liu, X.; et al. The Aegilops tauschii genome reveals multiple impacts of transposons. Nat. Plants 2017, 3, 946–955. [Google Scholar] [CrossRef]
- Jia, J.; Zhao, S.; Kong, X.; Li, Y.; Zhao, G.; He, W.; Appels, R.; Pfeifer, M.; Tao, Y.; Zhang, X.; et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 2013, 496, 91–95. [Google Scholar] [CrossRef]
- Levy, A.A.; Feldman, M. Evolution and origin of bread wheat. Plant Cell 2022, 34, 2549–2567. [Google Scholar] [CrossRef]
- Crespi, B.; Nosil, P. Conflictual speciation: Species formation via genomic conflict. Trends Ecol. Evol. 2013, 28, 48–57. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The Role of Transposable Elements in Speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Satish, L.; Kalendar, R.; Narayanan, M.; Kandasamy, S.; Sharma, A.; Emamverdian, A.; Wei, Q.; Zhou, M. The Dynamism of Transposon Methylation for Plant Development and Stress Adaptation. Int. J. Mol. Sci. 2021, 22, 11387. [Google Scholar] [CrossRef] [PubMed]
- Parisod, C.; Alix, K.; Just, J.; Petit, M.; Sarilar, V.; Mhiri, C.; Ainouche, M.; Chalhoub, B.; Grandbastien, M.A. Impact of transposable elements on the organization and function of allopolyploid genomes. New Phytol. 2010, 186, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Cho, J. Transposon-Derived Non-coding RNAs and Their Function in Plants. Front. Plant Sci. 2018, 9, 600. [Google Scholar] [CrossRef]
- Song, X.; Cao, X. Transposon-mediated epigenetic regulation contributes to phenotypic diversity and environmental adaptation in rice. Curr. Opin. Plant Biol. 2017, 36, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, L.J.; Joynson, R.; Omony, J.; Rusholme-Pilcher, R.; Olohan, L.; Lang, D.; Bai, C.; Hawkesford, M.; Salt, D.; Spannagl, M.; et al. Hidden variation in polyploid wheat drives local adaptation. Genome Res. 2018, 28, 1319–1332. [Google Scholar] [CrossRef]
- Shao, C.; Sun, S.; Liu, K.; Wang, J.; Li, S.; Liu, Q.; Deagle, B.E.; Seim, I.; Biscontin, A.; Wang, Q.; et al. The enormous repetitive Antarctic krill genome reveals environmental adaptations and population insights. Cell 2023, 186, 1279–1294 e19. [Google Scholar] [CrossRef]
- Yuan, J.; Sun, H.; Wang, Y.; Li, L.; Chen, S.; Jiao, W.; Jia, G.; Wang, L.; Mao, J.; Ni, Z.; et al. Open chromatin interaction maps reveal functional regulatory elements and chromatin architecture variations during wheat evolution. Genome Biol. 2022, 23, 34. [Google Scholar] [CrossRef]
- Wang, M.; Li, Z.; Zhang, Y.; Zhang, Y.; Xie, Y.; Ye, L.; Zhuang, Y.; Lin, K.; Zhao, F.; Guo, J.; et al. An atlas of wheat epigenetic regulatory elements reveals subgenome divergence in the regulation of development and stress responses. Plant Cell 2021, 33, 865–881. [Google Scholar] [CrossRef]
- Li, Z.; Wang, M.; Lin, K.; Xie, Y.; Guo, J.; Ye, L.; Zhuang, Y.; Teng, W.; Ran, X.; Tong, Y.; et al. The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. Genome Biol. 2019, 20, 139. [Google Scholar] [CrossRef]
- Keidar, D.; Doron, C.; Kashkush, K. Genome-wide analysis of a recently active retrotransposon, Au SINE, in wheat: Content, distribution within subgenomes and chromosomes, and gene associations. Plant Cell Rep. 2018, 37, 193–208. [Google Scholar] [CrossRef]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; De Oliveira, R.; International Wheat Genome Sequencing, C.; Mayer, K.F.X.; Paux, E.; et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, G.; Yang, Q.; Gao, L.; Liu, C.; Ru, Z.; Wang, D.; Jia, J.; Cui, D. Helitron and CACTA DNA transposons actively reshape the common wheat—AK58 genome. Genomics 2022, 114, 110288. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Vanzetti, L.S.; Sumner, A.; Dubcovsky, M.; Matvienko, M.; Distelfeld, A.; Michelmore, R.W.; Dubcovsky, J. Small RNAs, DNA methylation and transposable elements in wheat. BMC Genom. 2010, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Kraitshtein, Z.; Yaakov, B.; Khasdan, V.; Kashkush, K. Genetic and epigenetic dynamics of a retrotransposon after allopolyploidization of wheat. Genetics 2010, 186, 801–812. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Jia, J.; Xie, Y.; Cheng, J.; Kong, C.; Wang, M.; Gao, L.; Zhao, F.; Guo, J.; Wang, K.; Li, G.; et al. Homology-mediated inter-chromosomal interactions in hexaploid wheat lead to specific subgenome territories following polyploidization and introgression. Genome Biol. 2021, 22, 26. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Liu, J.; Zhang, Y.; Ye, L.; Peng, Y.; Wang, H.; Diao, H.; Ma, Y.; Wang, M.; et al. Transposable elements orchestrate subgenome-convergent and -divergent transcription in common wheat. Nat. Commun. 2022, 13, 6940. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, J.; Jia, G.; Ye, W.; Jeffrey Chen, Z.; Song, Q. Histone H3K27 dimethylation landscapes contribute to genome stability and genetic recombination during wheat polyploidization. Plant J. 2021, 105, 678–690. [Google Scholar] [CrossRef]
- Lu, F.H.; McKenzie, N.; Gardiner, L.J.; Luo, M.C.; Hall, A.; Bevan, M.W. Reduced chromatin accessibility underlies gene expression differences in homologous chromosome arms of diploid Aegilops tauschii and hexaploid wheat. Gigascience 2020, 9, giaa070. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, Y.; Kong, C.; Lu, Z.; Jia, H.; Ma, Z.; Zhang, Y.; Cui, D.; Ru, Z.; Wang, Y.; et al. Centromere repositioning and shifts in wheat evolution. Plant Commun. 2023, 100556. [Google Scholar] [CrossRef]
- Guo, X.; Han, F. Asymmetric epigenetic modification and elimination of rDNA sequences by polyploidization in wheat. Plant Cell 2014, 26, 4311–4327. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, W.; Wang, F.; Wu, X.; Wang, J. Asymmetric epigenetic modification and homoeolog expression bias in the establishment and evolution of allopolyploid Brassica napus. New Phytol. 2021, 232, 898–913. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Zhang, Y.; Lin, K.; Peng, Y.; Ye, L.; Zhuang, Y.; Wang, M.; Xie, Y.; Guo, J.; et al. Evolutionary rewiring of the wheat transcriptional regulatory network by lineage-specific transposable elements. Genome Res. 2021, 31, 2276–2289. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Defining an epigenetic code. Nat. Cell Biol. 2007, 9, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Li, B.; Choulet, F.; Heng, Y.; Hao, W.; Paux, E.; Liu, Z.; Yue, W.; Jin, W.; Feuillet, C.; Zhang, X. Wheat centromeric retrotransposons: The new ones take a major role in centromeric structure. Plant J. 2013, 73, 952–965. [Google Scholar] [CrossRef]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenet. Chromatin 2014, 7, 33. [Google Scholar] [CrossRef]
- Vera, D.L.; Madzima, T.F.; Labonne, J.D.; Alam, M.P.; Hoffman, G.G.; Girimurugan, S.B.; Zhang, J.; McGinnis, K.M.; Dennis, J.H.; Bass, H.W. Differential nuclease sensitivity profiling of chromatin reveals biochemical footprints coupled to gene expression and functional DNA elements in maize. Plant Cell 2014, 26, 3883–3893. [Google Scholar] [CrossRef]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 166, 1598. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef]
- Yan, W.; Chen, D.; Schumacher, J.; Durantini, D.; Engelhorn, J.; Chen, M.; Carles, C.C.; Kaufmann, K. Dynamic control of enhancer activity drives stage-specific gene expression during flower morphogenesis. Nat. Commun. 2019, 10, 1705. [Google Scholar] [CrossRef] [PubMed]
- Shewry, P.R.; Hey, S.J. The contribution of wheat to human diet and health. Food Energy Secur. 2015, 4, 178–202. [Google Scholar] [CrossRef] [PubMed]
- Scofield, S.R.; Huang, L.; Brandt, A.S.; Gill, B.S. Development of a virus-induced gene-silencing system for hexaploid wheat and its use in functional analysis of the Lr21-mediated leaf rust resistance pathway. Plant Physiol. 2005, 138, 2165–2173. [Google Scholar] [CrossRef]
- Bapela, T.; Shimelis, H.; Tsilo, T.J.; Mathew, I. Genetic Improvement of Wheat for Drought Tolerance: Progress, Challenges and Opportunities. Plants 2022, 11, 1331. [Google Scholar] [CrossRef]
- Xiong, W.; Reynolds, M.P.; Crossa, J.; Schulthess, U.; Sonder, K.; Montes, C.; Addimando, N.; Singh, R.P.; Ammar, K.; Gerard, B.; et al. Increased ranking change in wheat breeding under climate change. Nat. Plants 2021, 7, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Concia, L.; Veluchamy, A.; Ramirez-Prado, J.S.; Martin-Ramirez, A.; Huang, Y.; Perez, M.; Domenichini, S.; Granados, N.R.Y.; Kim, S.; Blein, T.; et al. Wheat chromatin architecture is organized in genome territories and transcription factories. Genome Biol. 2020, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, T.; Zhang, K.; You, Q.; Yan, H.; Zhao, N.; Yi, X.; Xu, W.; Su, Z. PCSD: A plant chromatin state database. Nucleic Acids Res. 2018, 46, D1157–D1167. [Google Scholar] [CrossRef]
- Roudier, F.; Ahmed, I.; Berard, C.; Sarazin, A.; Mary-Huard, T.; Cortijo, S.; Bouyer, D.; Caillieux, E.; Duvernois-Berthet, E.; Al-Shikhley, L.; et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011, 30, 1928–1938. [Google Scholar] [CrossRef]
- Du, Z.; Li, H.; Wei, Q.; Zhao, X.; Wang, C.; Zhu, Q.; Yi, X.; Xu, W.; Liu, X.S.; Jin, W.; et al. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol. Plant 2013, 6, 1463–1472. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Pfluger, J.; Wagner, D. Histone modifications and dynamic regulation of genome accessibility in plants. Curr. Opin. Plant Biol. 2007, 10, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.; Yuan, J.; Jiang, S.; Liu, Y.; Wang, L.; Liu, M.; Zheng, D.; Ye, W.; Wang, X.; Chen, Z.J. Asymmetrical changes of gene expression, small RNAs and chromatin in two resynthesized wheat allotetraploids. Plant J. 2018, 93, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Lin, Y.; Li, Y.; Wang, G. Dynamics of H3K27me3 Modification on Plant Adaptation to Environmental Cues. Plants 2021, 10, 1165. [Google Scholar] [CrossRef] [PubMed]
- Gorkin, D.U.; Barozzi, I.; Zhao, Y.; Zhang, Y.; Huang, H.; Lee, A.Y.; Li, B.; Chiou, J.; Wildberg, A.; Ding, B.; et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 2020, 583, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Jin, P.; Cui, X.; Gu, L.; Lu, Z.; Xue, Y.; Wei, L.; Qi, J.; Song, X.; Luo, M.; et al. Control of transposon activity by a histone H3K4 demethylase in rice. Proc. Natl. Acad. Sci. USA 2013, 110, 1953–1958. [Google Scholar] [CrossRef]
- Luo, C.; Sidote, D.J.; Zhang, Y.; Kerstetter, R.A.; Michael, T.P.; Lam, E. Integrative analysis of chromatin states in Arabidopsis identified potential regulatory mechanisms for natural antisense transcript production. Plant J. 2013, 73, 77–90. [Google Scholar] [CrossRef]
- Mhiri, C.; Borges, F.; Grandbastien, M.A. Specificities and Dynamics of Transposable Elements in Land Plants. Biology 2022, 11, 488. [Google Scholar] [CrossRef]
- Lu, Z.; Marand, A.P.; Ricci, W.A.; Ethridge, C.L.; Zhang, X.; Schmitz, R.J. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 2019, 5, 1250–1259. [Google Scholar] [CrossRef]
- Wessler, S.R. Phenotypic diversity mediated by the maize transposable elements Ac and Spm. Science 1988, 242, 399–405. [Google Scholar] [CrossRef]
- Hehl, R.; Nacken, W.K.; Krause, A.; Saedler, H.; Sommer, H. Structural analysis of Tam3, a transposable element from Antirrhinum majus, reveals homologies to the Ac element from maize. Plant Mol. Biol. 1991, 16, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Rommens, C.M.; van Haaren, M.J.; Nijkamp, H.J.; Hille, J. Differential repair of excision gaps generated by transposable elements of the ‘Ac family’. Bioessays 1993, 15, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; Ohme-Takagi, M.; Sarai, A. Unique mode of GCC box recognition by the DNA-binding domain of ethylene-responsive element-binding factor (ERF domain) in plant. J. Biol. Chem. 1998, 273, 26857–26861. [Google Scholar] [CrossRef] [PubMed]
- Ricci, W.A.; Lu, Z.; Ji, L.; Marand, A.P.; Ethridge, C.L.; Murphy, N.G.; Noshay, J.M.; Galli, M.; Mejia-Guerra, M.K.; Colome-Tatche, M.; et al. Widespread long-range cis-regulatory elements in the maize genome. Nat. Plants 2019, 5, 1237–1249. [Google Scholar] [CrossRef]
- Zhang, A.; Zhang, W. Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza sativa). Int. J. Mol. Sci. 2022, 23, 8947. [Google Scholar] [CrossRef]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef]
- Rodgers-Melnick, E.; Vera, D.L.; Bass, H.W.; Buckler, E.S. Open chromatin reveals the functional maize genome. Proc. Natl. Acad. Sci. USA 2016, 113, E3177–E3184. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004, 5, 276–287. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Annotation | Length (bp) | Percent (%) | Summary (%) |
|---|---|---|---|---|
| S1 | TE region 1 | 1,227,693,800 | 30.65 | S1–S9 (92.02) |
| S2 | TE region 2 | 314,684,600 | 7.86 | |
| S3 | TE region 3 | 72,102,000 | 1.80 | |
| S4 | TE region 4 | 136,383,200 | 3.40 | |
| S5 | TE region 5 (Unmarked) | 1,494,160,200 | 37.30 | |
| S6 | TE region 6 | 74,273,600 | 1.85 | |
| S7 | TE region 7 | 281,086,000 | 7.02 | |
| S8 | TE region 8 | 60,162,800 | 1.50 | |
| S9 | TE region 9 (H3K18ac-assosciated) | 25,231,000 | 0.63 | |
| S10 | H3K27me3 Polycomb | 125,719,600 | 3.14 | S10–S13 (5.91) |
| S11 | Bivalent state | 53,689,600 | 1.34 | |
| S12 | FLanking TSS | 26,332,400 | 0.66 | |
| S13 | Active TSS | 31,050,800 | 0.78 | |
| S14 | Intragenic region | 66,268,600 | 1.65 | 1.65 |
| S15 | Centromeric region | 16,660,000 | 0.42 | 0.42 |
| total | # | 4,005,498,200 | 100.00 | 100.00 |
| Distal TE-HSs | Proximal TE-HSs | ||
|---|---|---|---|
| TE Superfamily | Count (Ratio/%) | TE Superfamily | Count (Ratio/%) |
| Unknown | 795 (31.3) | Unknown | 160 (57.1) |
| LTR/Gypsy | 701 (27.6) | LINE/L1 | 34 (12.1) |
| LTR/Copia | 357 (14.1) | DNA/hAT-Ac | 29 (10.4) |
| LINE/L1 | 216 (8.5) | LTR/Gypsy | 19 (6.8) |
| DNA/CACTA | 161 (6.3) | LTR/Copia | 8 (2.9) |
| LTR retrotransposon | 114 (4.5) | DNA/CACTA | 7 (2.5) |
| DNA/CMC-EnSpm | 79 (3.1) | DNA/CMC-EnSpm | 5 (1.8) |
| DNA/hAT-Ac | 33 (1.3) | DNA/PIF-Harbinger | 5 (1.8) |
| DNA/MULE-MuDR | 17 (0.7) | DNA/MULE-MuDR | 3 (1.1) |
| DNA/PIF-Harbinger | 15 (0.6) | LINE/R1 | 2 (0.7) |
| LINE | 12 (0.5) | SINE/L1 | 2 (0.7) |
| DNA/hAT-Tip100 | 5 (0.2) | LTR/Pao | 1 (0.4) |
| DNA/PIF | 5 (0.2) | LTR/retrotransposon | 1 (0.4) |
| DNA/transposon | 4 (0.2) | DNA/hAT-Tag1 | 1 (0.4) |
| SINE/L1 | 4 (0.2) | DNA/Mutator | 1 (0.4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, C.; Zhao, G.; Gao, L.; Kong, X.; Wang, D.; Liu, X.; Jia, J. Epigenetic Landscape Is Largely Shaped by Diversiform Transposons in Aegilops tauschii. Int. J. Mol. Sci. 2023, 24, 9349. https://doi.org/10.3390/ijms24119349
Kong C, Zhao G, Gao L, Kong X, Wang D, Liu X, Jia J. Epigenetic Landscape Is Largely Shaped by Diversiform Transposons in Aegilops tauschii. International Journal of Molecular Sciences. 2023; 24(11):9349. https://doi.org/10.3390/ijms24119349
Chicago/Turabian StyleKong, Chuizheng, Guangyao Zhao, Lifeng Gao, Xiuying Kong, Daowen Wang, Xu Liu, and Jizeng Jia. 2023. "Epigenetic Landscape Is Largely Shaped by Diversiform Transposons in Aegilops tauschii" International Journal of Molecular Sciences 24, no. 11: 9349. https://doi.org/10.3390/ijms24119349
APA StyleKong, C., Zhao, G., Gao, L., Kong, X., Wang, D., Liu, X., & Jia, J. (2023). Epigenetic Landscape Is Largely Shaped by Diversiform Transposons in Aegilops tauschii. International Journal of Molecular Sciences, 24(11), 9349. https://doi.org/10.3390/ijms24119349