
Mitogenome Characterization of Four Conus Species and Comparative Analysis

Abstract
:1. Introduction
2. Results
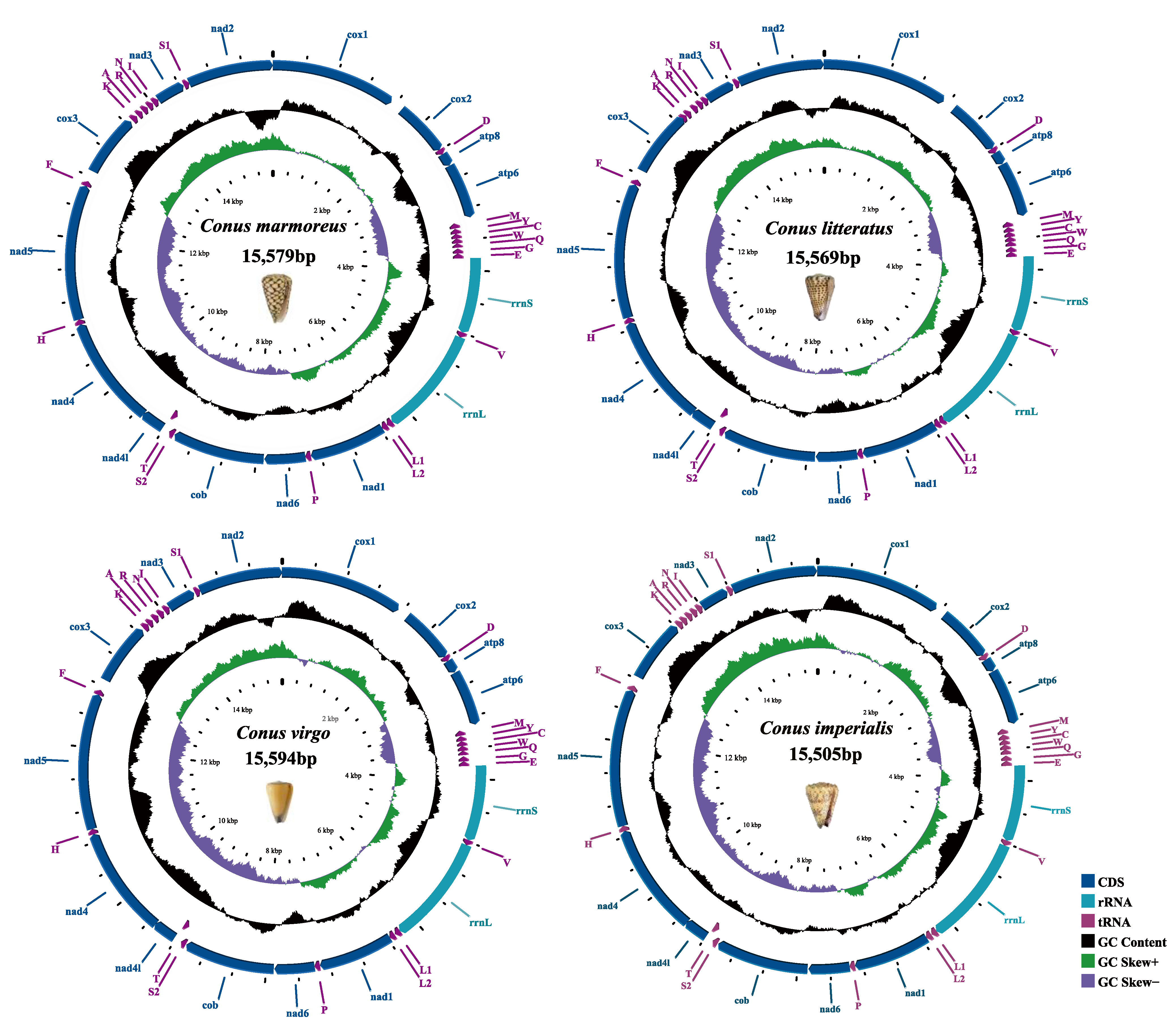
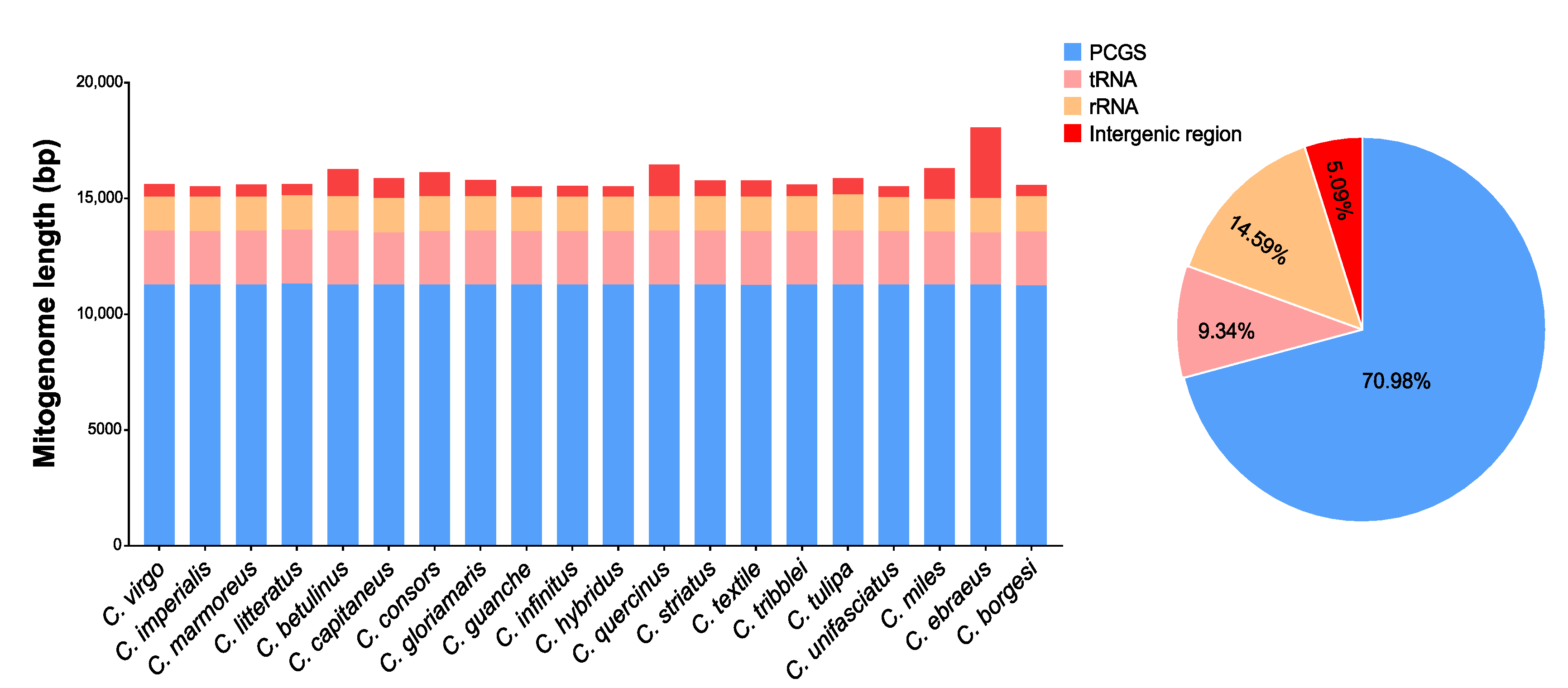
2.1. Genome Structure and Organization
Nucleotide Composition
2.2. Protein-Coding Genes Analysis
Codon Preference Statistics
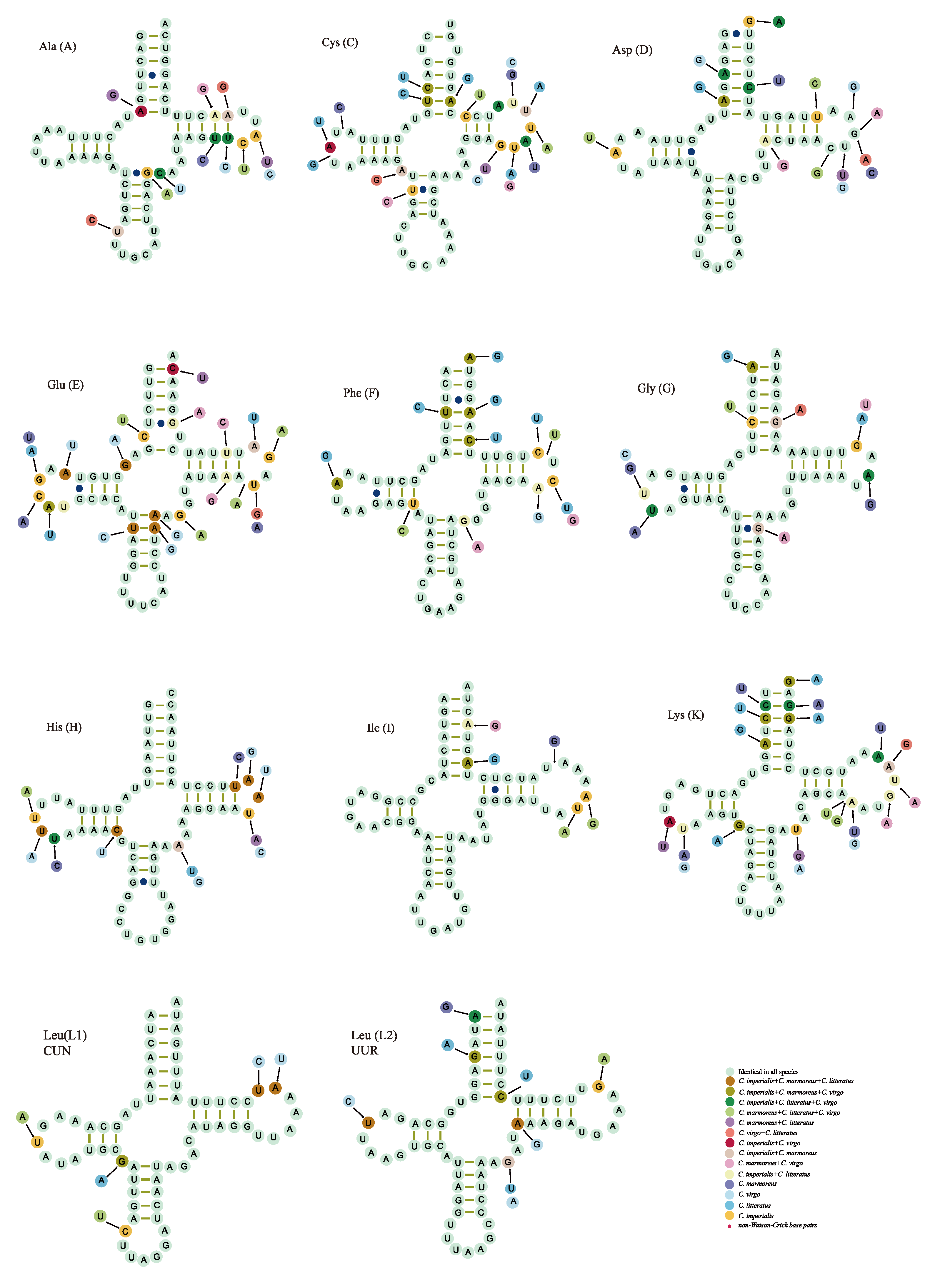
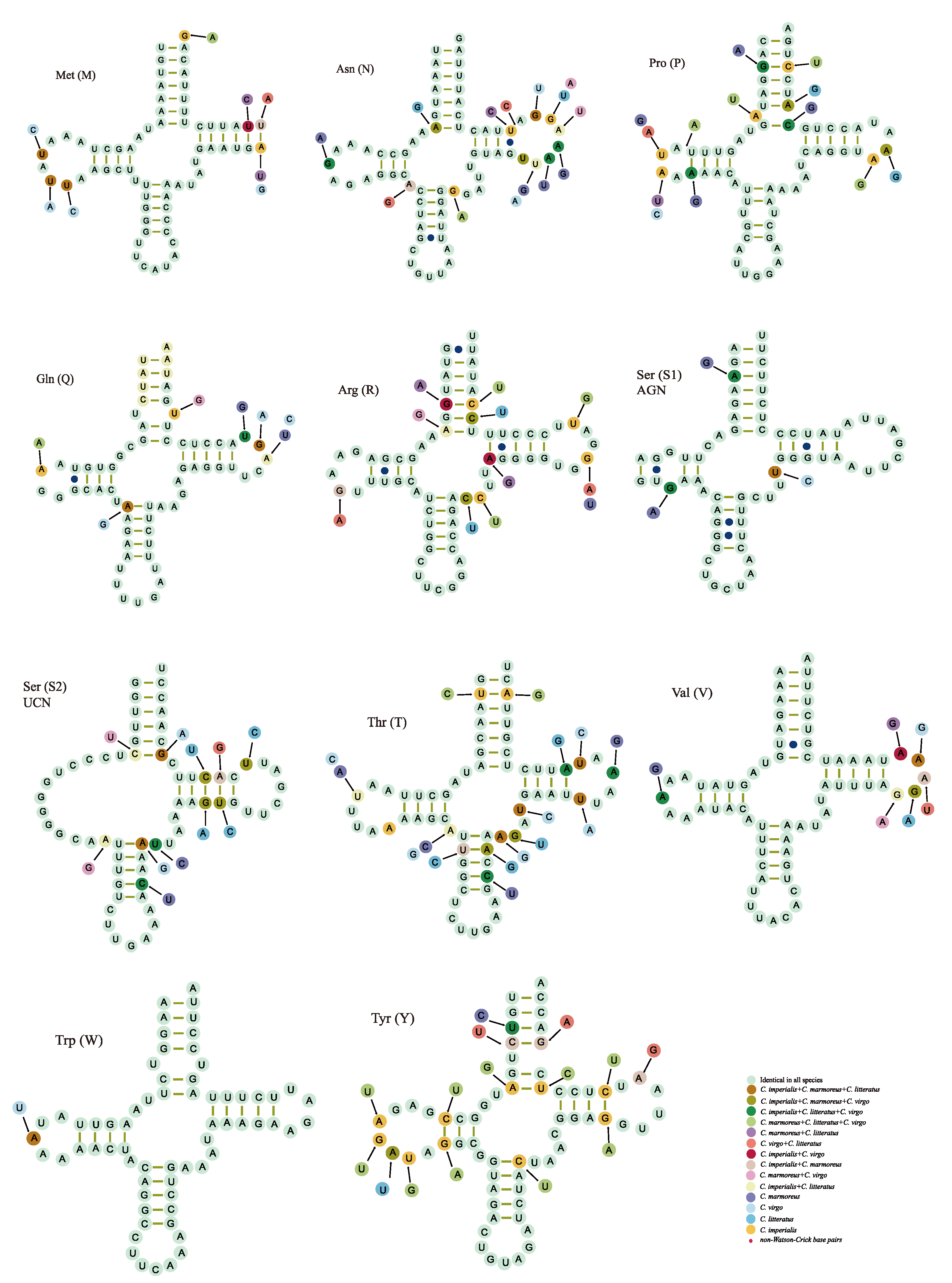
2.3. Transfer RNA Genes
2.4. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Specimen Collection and DNA Extraction
4.2. PCR Amplification and Sequencing
4.3. Genome Assembly and Annotation
4.4. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, S.X.; Dong, Q.X.; Shen, J.H.; Yu, X.J.; Du, H.; Huang, C.J. Imposex of Conus betulinus and Conus vexillum. Acta Ecol. Sin. 2005, 25, 289–297. [Google Scholar]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Yang, Y.; Tan, Y.; Zhangsun, D.; Zhu, X.; Luo, S. Design, Synthesis, and Activity of an α-Conotoxin LtIA Fluorescent Analogue. ACS Chem. Neurosci. 2021, 12, 3662–3671. [Google Scholar] [CrossRef]
- Pan, S.; Fan, Y.; Zhu, X.; Xue, Y.; Luo, S.; Wang, X. From Crystal Structures of RgIA4 in Complex with Ac-AChBP to Molecular Determinants of Its High Potency of α9α10 nAChR. Mar. Drugs 2021, 19, 709. [Google Scholar] [CrossRef]
- Li, X.; Xiong, J.; Zhang, B.; Zhangsun, D.; Luo, S. α-Conotoxin TxIB Inhibits Development of Morphine-Induced Conditioned Place Preference in Mice via Blocking α6β2* Nicotinic Acetylcholine Receptors. Front. Pharmacol. 2021, 12, 772990. [Google Scholar] [CrossRef]
- Puillandre, N.; Bouchet, P.; Duda, T.F., Jr.; Kauferstein, S.; Kohn, A.J.; Olivera, B.M.; Watkins, M.; Meyer, C. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogen Evol. 2014, 78, 290–303. [Google Scholar] [CrossRef]
- Puillandre, N.; Holford, M. The Terebridae and teretoxins: Combining phylogeny and anatomy for concerted discovery of bioactive compounds. BMC Chem. Biol. 2010, 10, 7–12. [Google Scholar] [CrossRef]
- Hillis, D.M.; Dixon, M.T. Ribosomal DNA: Molecular evolution and phylogenetic inference. Q. Rev. Biol. 1991, 66, 411–453. [Google Scholar] [CrossRef]
- Puillandre, N.; Samadi, S.; Boisselier, M.C.; Sysoev, A.V.; Kantor, Y.I.; Cruaud, C.; Couloux, A.; Bouchet, P. Starting to unravel the toxoglossan knot: Molecular phylogeny of the “turrids” (Neogastropoda: Conoidea). Mol. Phylogenet. Evol. 2008, 47, 1122–1134. [Google Scholar] [CrossRef]
- Kraus, N.J.; Corneli, P.S.; Watkins, M.; Bandyopadhyay, P.K.; Seger, J.; Olivera, B.M. Against expectation: A short sequence with high signal elucidates cone snail phylogeny. Mol. Phylogenet. Evol. 2011, 58, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Lawler, A.J.; Duda, T.F. Molecular and morphometric data suggest the presence of a neglected species in the marine gastropod family Conidae. Mol. Phylogenet. Evol. 2017, 109, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Uribe, J.E.; Zardoya, R.; Puillandre, N. Phylogenetic relationships of the conoidean snails (Gastropoda: Caenogastropoda) based on mitochondrial genomes. Mol. Phylogenet. Evol. 2018, 127, 898–906. [Google Scholar] [CrossRef]
- Chen, P.W.; Hsiao, S.T.; Huang, C.W.; Chen, K.S.; Tseng, C.T.; Wu, W.L.; Hwang, D.F. The complete mitochondrial genome of Conus tulipa (Neogastropoda: Conidae). Mitochondrial DNA Part A 2016, 27, 2738–2739. [Google Scholar] [CrossRef]
- Uribe, J.E.; Puillandre, N.; Zardoya, R. Beyond Conus: Phylogenetic relationships of Conidae based on complete mitochondrial genomes. Mol. Phylogenet. Evol. 2017, 107, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.L.; Zardoya, R. Mitogenomic phylogeny of cone snails endemic to Senegal. Mol. Phylogenet. Evol. 2017, 112, 79–87. [Google Scholar] [CrossRef]
- WoRMS Editorial Board. World Register of Marine Species. Available online: http://www.marinespecies.org (accessed on 7 December 2019).
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta BBA Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, X.; Sha, Z.; Yan, B.; Xu, Q. Complete mitochondrial genome of the Japanese snapping shrimp Alpheus japonicus (Crustacea: Decapoda: Caridea): Gene rearrangement and phylogeny within Caridea. Sci. China Life Sci. 2012, 55, 591–598. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Sun, S.; Kong, L.; Yu, H.; Li, Q. The complete mitochondrial DNA of Tegillarca granosa and comparative mitogenomic analyses of three Arcidae species. Gene 2015, 557, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, H.; Steiner, G. The complete sequence and gene organization of the mitochondrial genome of the gadilid scaphopod Siphonondentalium lobatum (Mollusca). Mol. Phylogenet. Evol. 2004, 31, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Tang, P.; Zheng, L.H.; Shi, M.; Chen, X.X. The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A+T content and a long intergenic spacer between atp8 and atp6. Mol. Biol. Rep. 2010, 37, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Varani, G.; McClain, W.H. The G·U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Shi, X.; Wu, Q.; Tao, Y.; Guo, H.; Ji, C.; Bai, Y. Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int. J. Biol. Macromol. 2018, 118, 31–40. [Google Scholar] [CrossRef]
- Zhang, B.; Havird, J.C.; Wang, E.; Lv, J.; Xu, X. Massive gene rearrangement in mitogenomes of phytoseiid mites. Int. J. Biol. Macromol. 2021, 186, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.L.; Uribe, J.E.; Echeverry, A.M.; Zardoya, R. Phylogenetic relationships of cone snails endemic to Cabo Verde based on mitochondrial genomes. BMC Evol. Biol. 2017, 17, 231. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Chen, Q.; Zhang, J.; Shi, Q. Mitochondrial genome sequencing of a vermivorous cone snail Conus quercinus supports the correlative analysis between phylogenetic relationships and dietary types of Conus species. PLoS ONE 2018, 13, e0193053. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.W.; Hsiao, S.T.; Chen, K.S.; Tseng, C.T.; Wu, W.L.; Hwang, D.F. The complete mitochondrial genome of Conus striatus (Neogastropoda: Conidae). Mitochondrial DNA Part B 2016, 1, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.W.; Hsiao, S.T.; Chen, K.S.; Tseng, C.T.; Wu, W.L.; Hwang, D.F. The complete mitochondrial genome of Conus capitaneus (Neogastropoda: Conidae). Mitochondrial DNA Part B 2016, 1, 520–521. [Google Scholar] [CrossRef] [PubMed]
- Barghi, N.; Concepcion, G.P.; Olivera, B.M.; Lluisma, A.O. Characterization of the complete mitochondrial genome of Conus tribblei Walls, 1977. Mitochondrial DNA Part A 2016, 27, 4451–4452. [Google Scholar] [CrossRef] [PubMed]
- Brauer, A.; Kurz, A.; Stockwell, T.; Baden-Tillson, H.; Heidler, J.; Wittig, I.; Kauferstein, S.; Mebs, D.; Stocklin, R.; Remm, M. The mitochondrial genome of the venomous cone snail Conus consors. PLoS ONE 2012, 7, e51528. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | A + T% | AT-Skew | GC-Skew | Species | A + T% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|
| C. virgo * | 64.42 | −0.17 | 0.15 | C. unifasciatus | 66.08 | −0.16 | 0.11 |
| C. imperialis * | 65.96 | −0.15 | 0.1 | C. borgesi | 67.15 | −0.15 | 0.11 |
| C. litteratus * | 66.05 | −0.16 | 0.12 | C. quercinus | 66.47 | −0.15 | 0.12 |
| C. marmoreus * | 66.81 | −0.15 | 0.12 | C. striatus | 64.54 | −0.2 | 0.17 |
| C. consors | 67.12 | −0.17 | 0.18 | C. tribblei | 65.95 | −0.15 | 0.1 |
| C. gloriamaris | 66.27 | −0.16 | 0.11 | C. tulipa | 66.4 | −0.14 | 0.1 |
| C. textile | 65.21 | −0.16 | 0.1 | C. ebraeus | 66.55 | −0.12 | 0.06 |
| C. guanche | 66.07 | −0.16 | 0.12 | C. betulinus | 63.93 | −0.2 | 0.19 |
| C. infinitus | 67.19 | −0.15 | 0.12 | C. capitaneus | 62.23 | −0.18 | 0.14 |
| C. hybridus | 66.04 | −0.16 | 0.11 | C. miles | 61.84 | −0.17 | 0.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhu, X.; Liu, Y.; Luo, S.; Zhangsun, D. Mitogenome Characterization of Four Conus Species and Comparative Analysis. Int. J. Mol. Sci. 2023, 24, 9411. https://doi.org/10.3390/ijms24119411
Wang H, Zhu X, Liu Y, Luo S, Zhangsun D. Mitogenome Characterization of Four Conus Species and Comparative Analysis. International Journal of Molecular Sciences. 2023; 24(11):9411. https://doi.org/10.3390/ijms24119411
Chicago/Turabian StyleWang, Hao, Xiaopeng Zhu, Yuepeng Liu, Sulan Luo, and Dongting Zhangsun. 2023. "Mitogenome Characterization of Four Conus Species and Comparative Analysis" International Journal of Molecular Sciences 24, no. 11: 9411. https://doi.org/10.3390/ijms24119411