1. Introduction
The protein-synthesizing machinery in all living cells is the ribosome. The ribosome is composed of 55 ribosomal proteins (RPs) and 3 ribosomal RNAs (rRNAs) in
Escherichia coli; in mammals, it is composed of 80 RPs and 4 rRNAs, while in budding yeast it is composed of 79 RPs and 4 rRNAs [
1,
2]. The transcription of ribosomal protein genes (RPGs) in all species is thought to be tightly controlled and coordinately expressed. In eukaryotes, these genes form a transcriptional module containing one or more common DNA motifs, which bind transcriptional activators [
3]. In
E. coli, the RPGs form an operon in which a single promoter controls the expression; however, in eukaryotes—such as yeasts, plants, and animals—regulation is much more complicated and less understood than in prokaryotes [
3]. In eukaryotes, the RPGs are scattered throughout the entire genome with no operon structure but, surprisingly, co-regulation and the coordinated expression are still observed [
3,
4]. Since this transcriptional module is small and the amino-acidic sequence of RPs is highly conserved, understanding the basic principles coordinating gene regulation of this transcriptional module will significantly contribute to the knowledge and function of other transcriptional modules.
The fission yeast genome contains 141 RPGs, encoding the complete set of 79 RPs, forming the ribosome with four rRNAs [
5,
6]. Since only 79 RPs form the ribosome and there are 141 RPGs, a single RP can be encoded for more than one gene, indicating that some RPGs have been duplicated and can constitute a family of RPGs [
5,
6]. Most RPs in plants and fungi are encoded for more than one RPG, while mammals and protists contain only a few duplicated RPGs [
7]. The duplicated genes are thought to have arisen from genome duplication, genome hybridization, retroduplication and, in some cases, polyploidy. The differences in the numbers of RPGs in these genomes are not related to genome size but, rather, are associated with the organism’s overall propensity to gene duplication [
8,
9].
The initial characterization of 14 promoters of RPGs in fission yeast showed discrete conserved modules, which were named the homology (Homol) A, B, C, D, and E regions [
5,
10,
11]. Surprisingly, these Homol regions were completely different from previously described promoter elements of budding yeast and mammals such as Rap1 (budding yeast), TATA box, initiator (Inr), downstream promoter element (DPE), TCT initiator, and motif 10 element (MTE). The function of each Homol region was studied using a promoter deletion approach in fission yeast. It was found that the HomolA, -B, and -C regions play a role in regulating transcription and might have an upstream activating sequence (UAS)-like function; moreover, they were not conserved in all of the RPG promoters [
5,
10,
11]. In contrast, the HomolD sequence was present in all of the studied RPG promoters, and it was able to direct the initiation of transcription in an analog fashion, similar to the TATA box [
5,
10]. This conserved sequence is the octamer CAGTCACA/G and is located 39–52 base pairs (bp) upstream of the transcription start site (TSS), in a position where the TATA box is usually located in fission yeast [
5,
10]. In some promoters, the inverted form TGTGACTG is found and is fully functional to direct the initiation of transcription. In an in vivo assay, using a reporter gene assay, it was demonstrated that the HomolD box is necessary and sufficient to direct and initiate transcription from the RPGs and, thus, can act as a TATA box analog. An electrophoretic mobility shift assay (EMSA) also found that the HomolD box binds a factor different from the TATA-box-binding protein (TBP). Point mutations in the HomolD box can completely abolish the ability of this DNA motif to direct the initiation of transcription from the RPGs [
5,
10].
RPs from fission yeast can be encoded by two or three duplicated RPGs, which contain a HomolD box in their promoters to initiate transcription. Interestingly, in each RPG family, at least one of those members possesses a sequence upstream of the HomolD box named HomolE box, which is a tandem repeated DNA motif with a consensus sequence of ACCCTACCCT or the inverted form AGGGTAGGGT [
5,
11]. This DNA motif corresponds to a proximal UAS-like element for HomolD-containing promoters, since this sequence upstream of the HomolD box strongly increases transcription in vivo [
5,
11]. The HomolE and HomolD can occur in those promoters in both orientations, and the distance between them varies from 0 to 32 bp [
5,
11]. Five size classes of spacers can be found setting the HomolE–HomolD promoter arrangement, which are 0–2, 6–8, 10–12, 14–17, and 21 bp apart [
5,
11]. The distance between both boxes is critical in the transcriptional activation of RPGs. It has been demonstrated that the smaller the distance between the HomolE and HomolD boxes, the higher the activity of the promoter arrangement [
5,
11]. The sequence context between HomolE and HomolD is also important for transcriptional activation. To be fully functional, both promoter elements must be in the same orientation as they are in natural promoters. For example, the arrangement AGGGTAGGGT-TGTGACTG found in a natural promoter is fully functional and strongly activates transcription; in contrast, the inverted arrangement ACCCTACCCT-CAGTCACA only has basal activity [
5,
11].
The fission yeast Schizosaccharomyces pombe is a valuable biological model for studying cellular processes, and genetic, biochemical, and bioinformatic tools can be applied. In this work, we identified a HomolE-binding protein from fission yeast and found that the recombinant protein can activate in vitro transcription in a cell-free extract using a synthetic promoter containing a HomolD box and two upstream HomolE boxes. The identified HomolE-binding protein (HEBP) is the product of the fhl1 gene.
3. Discussion
In this work, we identified an HEBP from fission yeast as the product of the fhl1 gene. The purified recombinant protein bound to the HomolE box and was able to activate transcription in a fission yeast WCE with a synthetic promoter containing both a HomolD box and upstream HomolE boxes, strongly suggesting that the product of the fhl1 gene is an HEBP. The HomolE box is always found upstream of HomolD-box-containing promoters, which are present in RPGs and several non-RPGs.
Ribosome biogenesis is one of the most energy-intensive anabolic processes carried out by growing cells and is highly regulated in response to growth and stress signals [
14,
15,
16]. It is estimated that in rapidly growing budding yeast, the ribosomes are produced at a rate of 2000 per minute and can be present at 200,000 copies per cell [
16]. This requires the dedication of almost 50% of all RNA polymerase II (RNAPII) initiation events in RPG transcription and ribosome biogenesis genes (RiBi genes), along with a high production of rRNAs, which are produced by the combined action of RNA polymerase I (RNAPI) and RNA polymerase III (RNAPIII) [
15,
16]. The production of RPs and rRNAs must be highly coordinated in order to ensure a rapid and efficient ribosome assembly. This coordination must begin at the transcription level, stimulated by environmental signals that favor rapid growth and are downregulated by stress conditions. However, RPGs’ coordinated regulation and expression with other ribosomal components are still poorly understood in fission yeast. DNA microarray studies have shown that all 141 RPGs have tightly coordinated expressions of those genes. For example, during the switch from vegetative to meiotic growth, the expression of the RPGs is downregulated; however, within a short time, they are strongly reactivated at the beginning of meiosis [
5,
17]. The co-expression profile can be seen in 32 of the 59 non-RPGs containing a HomolD box, indicating that this DNA motif is responsible for the coordinated expression of RPGs and non-RPGs displaying a HomolD box in their promoters [
5,
17].
As stated earlier, the HomolE box has always been found upstream of the HomolD box in RPGs and in non-RPGs, which are transcribed by RNAPII [
18]. The HomolD box binds a protein factor with biochemical features that are different from those of TBP. In an attempt to biochemically analyze the factor that binds to the HomolD box, we used DNA affinity chromatography linking a double-stranded multimerized HomolD sequence to the matrix to isolate the factor from a partially purified fraction of cell extract enriched in HomolD-binding activity [
18]. Bound proteins were eluted from the affinity column and analyzed by mass spectrometry. The results indicated that the RNAPI transcription factor Rrn7 was identified in the bound fraction from the HomolD-box affinity column, which is required for RPG transcription [
18]. The Rrn7 transcription factor is also a member of the RNAPI transcription machinery and is involved in the transcription of rRNA [
18,
19]. This factor binds to a conserved sequence in the rRNA gene, similar to the HomolD box [
18,
19]. However, the RPGs are transcribed by the RNAPII transcription apparatus, since the transcription in vitro is sensitive to α-amanitin [
18]. This factor is a member of the Zn-ribbon protein family related to the RNAPII transcription factor TFIIB, which plays a pivotal role in the preinitiation complex (PIC) assembly on eukaryotic protein-coding genes [
20,
21]. Rrn7 possesses a Zn-ribbon at the N-terminus of the polypeptide and two cyclin fold domains at the C-terminus of the protein, and it displays domain conservation with the TFIIB family members [
18,
19,
20,
21]. The Rrn7 is required for transcription of the RPGs’ HomolD-box-containing promoters and, most likely, for the non-RPGs containing a HomolD box as well. This transcription factor is encoded by an essential gene for viability (Pombase), and unfortunately, no temperature-sensitive alleles have been isolated to study its in vivo function deeply. The HEBP (Fhl1 protein) probably interacts with Rrn7 to activate transcription or, alternatively, the Fhl1 protein interacts with the mediator complex or other components of the RNAPII transcription machinery.
Despite the conservation of the coding sequences of the RPGs from different eukaryotes, the promoter sequences controlling their expression are not conserved, and different cis-elements can be found in the different species. Therefore, it is necessary to compare the RPG promoters from different genomes and identify conserved cis-elements associated with them in each species. It is also necessary to identify the trans-acting protein factors that bind to each conserved DNA motif in the RPG promoters, and to study the molecular mechanisms associated with their expression. The transcriptional control of the RPGs is well known in budding yeast but differs from the transcriptional control of RPGs in fission yeast and mammalian cells [
3,
6,
16]. In budding yeast cells, three distinct RPG promoter architectures lead to an extensive—albeit incomplete—transcriptional co-regulation [
16]. Those promoter architectures have been named Category I, II, and III. Most of the promoters in budding yeast are bound by the Rap1 protein (127 out of 138), and this factor can be found in promoters of Category I and Category II promoters [
16]. However, Rap1 also binds to a large number of non-RPG promoters [
22]. Furthermore, the transcription factor FHL1 can interact and recruit IFH1 (interacts with fork-head 1); both are found at RPG promoters in budding yeast, and they are highly specific for those genes, with very few non-RPG binding sites [
16,
23,
24,
25,
26]. Fork-head transcription factors (also known as winged-helix transcription factors) constitute a family of sequence-specific regulators possessing a conserved DNA-binding domain—a variant of the helix-turn-helix domain. They are important in controlling many cellular and developmental processes in eukaryotes. Coding genes for fork-head transcription factors have been described for budding and fission yeasts, among other organisms. Four genes encoding fork-head transcription factors have been identified in fission yeast:
mei4,
sep1,
fkh2, and
fhl1 [
27,
28].
The fission yeast
fhl1 gene has not yet been thoroughly characterized. It has been suggested that
fhl1 could regulate sporulation, typically induced under poor nutrient conditions [
27]. Deletion of
fhl1 causes a reduced cell growth rate and an extension of cell length due to delayed G2-to-M transition. The mutant deletion strain cells are more resistant to killing by the mutagen methyl methane sulfonate than wild-type cells [
27]. The
fhl1 gene is not essential for cell viability, but mutant cells have longer doubling times, and the homothallic cells show stronger sporulation [
27,
28]. Genome-wide gene expression analysis of the
fhl1 mutant strain has revealed that the expression of several genes is dysregulated. Those dysregulated genes in the mutant strain belong to the nitrogen starvation response, mating, sporulation, nutrient sensing, transport, and permease genes [
28]. Tor2 also regulates some of the genes regulated by Fhl1, and it might be possible that Fhl1 acts in TOR signaling downstream of Tor2 [
28]. The Fhl1 protein may be regulated by phosphorylation by Tor2 or, alternatively, by a protein kinase regulated by Tor2. Moreover, it has been demonstrated that fission yeast Fhl1 regulates only a few RPGs and stress-response genes, in contrast to the budding yeast
FHL1 gene [
28,
29,
30]. Only two RPGs were found to be downregulated in the
fhl1 mutant strain—namely,
rpl603 and
rps2202, of which
rps2202 possesses a HomolE box in its promoter. Perhaps the role of Fhl1 is to regulate transcription from HomolE-containing promoters under certain physiological conditions in fission yeast. Notably, promoters containing only a HomolD box have basal transcriptional activity, which could explain why the HomolE box is unnecessary for the RPGs to be transcribed [
5,
11,
18].
The Fhl1 protein has FHA and FH domains, similar to the budding yeast FHL1 protein homolog. The FHA domain might be important to interact with other protein factors involved in transcription, while the FH domain might bind DNA. We did not notice any other important domain in this protein. Additional biochemical work is needed to define the interacting, DNA-binding, and activation domains in the Fhl1 protein. Furthermore, genetic work is necessary to confirm in vivo that the fhl1 gene regulates the RPGs containing HomolE boxes.
4. Materials and Methods
4.1. Purification of HomolE-Binding Protein
The HEBP was purified from the wild-type fission yeast strain 972h-. Whole-cell extracts (WCEs) were prepared from 100 g (wet weight) of cells by grinding the cells with a mortar and liquid nitrogen, as described by Rojas et al. [
18]. The obtained WCE (100 mL at 10 mg/mL protein concentration) was loaded onto a 50 mL phosphocellulose (Whatman P11) column that had previously been equilibrated with buffer A (20 mM HEPES pH 7.9, 50 mM KCl, 0.5 mM EDTA, 2.5 mM DTT, 0.5 mM PMSF, and 10%
v/
v glycerol). After protein loading, the column was washed extensively with buffer A and eluted stepwise with 0.2 M, 0.5 M, and 1.0 M KCl in buffer A. The HEBP activity was determined by electrophoretic mobility shift assay (EMSA), and the binding activity was found in the 0.5 M KCl fraction from the P11 column. This protein fraction (200 mg) was dialyzed against buffer A and loaded onto an S-Sepharose column (20 mL column). Bound proteins were eluted with a linear KCl gradient (0.05 to 0.5 M) in buffer A. Fractions containing HEBP activity were pooled (50 mg) and dialyzed against buffer B (20 mM sodium phosphate pH 7.9, 2.5 mM DTT, 0.5 mM PMSF, and 10%
v/
v glycerol) and loaded onto a hydroxyapatite column (10 mL) equilibrated in buffer B. The bound proteins were eluted with a linear gradient of sodium phosphate buffer (0.05–0.25 M) in buffer B. Those fractions containing HEBP activity (5 mg) were pooled and dialyzed against buffer A and loaded onto a Poros-S column (1 mL), and then eluted with a linear gradient of KCl (0.05–0.5 M) in buffer A. HEBP-binding activity was eluted as a sharp peak around a 0.25 M KCl concentration. The fractions containing HEBP activity were pooled and analyzed by SDS-PAGE, followed by silver staining, or analyzed by Southwestern blot, as described below.
4.2. Electrophoretic Mobility Shift Assay
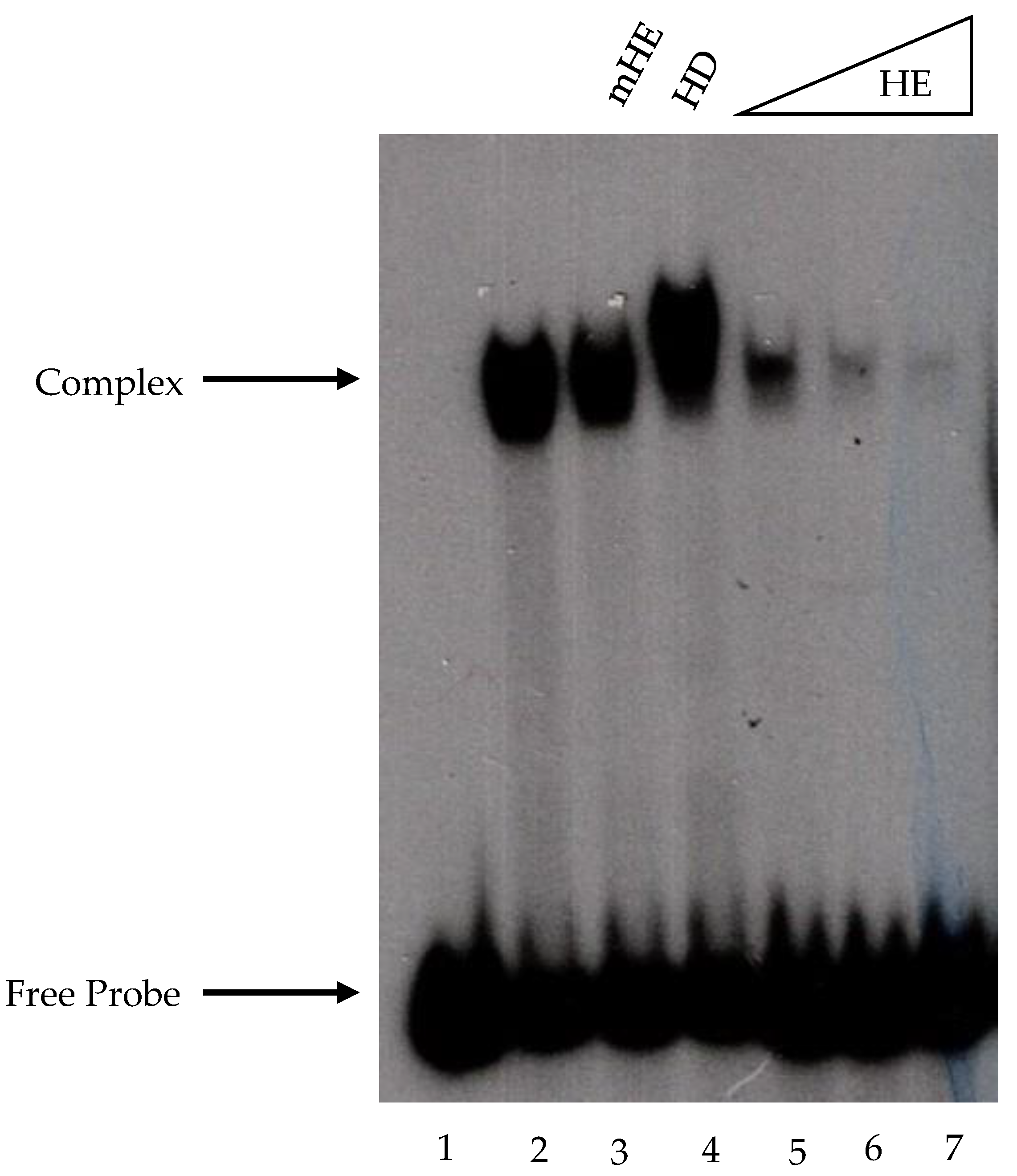
EMSA was used to monitor the HEBP activity from the different chromatographic steps. The EMSA was performed as described previously [
18]. Each binding reaction contained a binding mix: 20 mM HEPES (pH 7.9), 50 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 5% glycerol, 0.5% PEG 8000 (Sigma-Aldrich, St. Louis, MO, USA), 5 mM DTT, 0.1 mM PMSF, 5 μg of acetylated BSA, and 0.5 μg of sonicated salmon sperm DNA. The proteins were incubated with the binding mix for 5 min at 25 °C. Then, 5–10 ng of two tandem double-stranded HomolE boxes containing the probe TTCGTTGAGGGTAGGGTTGAGGGTAGGGTTATGC, end-labeled by T4 polynucleotide kinase and
32γATP, was added to the assays, and the reaction mixes were incubated for 15 min at 30 °C. The DNA–protein complexes were evaluated in 5% polyacrylamide gels containing 2% glycerol and run at 100 V and 4 °C for 2 h in Tris-borate-EDTA (pH 8.3) buffer. Complex detection was performed by autoradiography analysis.
The EMSA performed with recombinant Fhl1 protein was carried out as described above, with slight modifications. The non-specific competitor salmon sperm DNA was replaced with 50 ng of poly (dG-dC), and the PEG was omitted. The polyacrylamide gels used to resolve the complexes contained 1 mM DTT.
4.3. In Vitro Transcription Assays
In vitro transcription was performed as described previously [
18], using 100 ng of HomolD-box- or HomolE–HomolD-box-containing templates. The synthetic promoter sequence of the HomolD box (shaded blue) from the rpK5 promoter was AAACAGTCACATTTTACAACAATTCACCACTTCAATTTCCAACCTCAATACCCTATTCTCCAACAACCAA.
The synthetic promoter sequence of the HomolE–HomolD construct was CGAATTCGTTGAGGGTAGGGTtgAGGGTAGGGTtgCAGTCACATTTTA CAACAATTCACCACTTCAATTTCCAACCTCAATACCCTATTCTCCAACAACCAA. This promoter contained 2 HomolE boxes (shaded green) separated by a 2 bp spacer (tg). The HomolE boxes are shaded green, and the HomolD box is shaded blue. The mutant HomolE box contained two base changes indicated in lowercase (AGtGTAGtGT). These promoters were fused to the G-minus cassette and, upon digestion of the transcripts by RNase T1, produced a transcript of 370 nucleotides in length. The reactions were performed with 5 μL of WCE (10 mg/mL). Transcript detection was performed by autoradiography analysis.
4.4. Southwestern Blot Analysis
Proteins were separated on an 8% SDS-PAGE and electro-transferred to an Immobilon-P membrane in 20 mM Tris, 200 mM glycine, and 20% v/v methanol. The membrane containing the proteins was incubated overnight at 8 °C in TNED buffer (10 mM Tris pH 7.9, 50 mM NaCl, 0.1 mM EDTA, and 1 mM DTT), supplemented with 2.5% BSA and 20 μg/mL sonicated salmon sperm DNA. Afterward, the membrane was incubated with 1 × 106 cpm/mL of a HomolE end-labeled probe (the same as used for EMSA) or a mutant HomolE end-labeled probe (TTCGTTGAGtGTAGtGTTGAGtGTAGtGTTAT GC) for 14 h at room temperature. After probe incubation, the membrane was washed four times for 20 min with TNED buffer and exposed to an X-ray film.
4.5. Recombinant fhl1 Protein Expression and Purification
The coding sequence of the fhl1 gene (SPAC1142.08) was synthesized at GenScript (Piscataway, NJ, USA) and inserted into the expression vector pET15b (Novagen, St. Louis, MO, USA). E. coli BL21 (DE3) cells were transformed with pET15b containing the corresponding gene sequences, grown in LB media until OD600 0.8. Then, the protein expression was induced with 0.5 mM IPTG (Calbiochem, St. Louis, MO, USA) to obtain appropriate amounts of the recombinant protein. To recover the recombinant protein, first, the cell pellet was washed with STE buffer (100 mM NaCl, 10 mM Tris HCl pH 8.0, 1 mM EDTA). Then, the cells were sonicated and centrifuged to collect the pellets, which were washed with lysis buffer (50 mM HEPES pH 7.9, 5% glycerol, 2 mM EDTA, 0.1 mM DTT, 0.05% DOC, 1% Triton X-100) and then washed again with the same buffer but without detergents. The washed pellet was mixed with 40 mL of guanidine buffer (6 M guanidine hydrochloride, 10 mM HEPES pH 7.9, 0.2 mM EDTA, 0.2 mM EGTA, 2 mM DTT) and was incubated overnight at 4 °C. The next day, the protein mix was diluted with 160 mL of dilution buffer (10 mM HEPES pH 7.9, 0.2 mM EDTA, 0.2 mM EGTA, 2 mM DTT) and incubated overnight at 4 °C. Then, the mix was dialyzed for 4 h at 4 °C against 20 mM Tris pH 7.5, 0.5 M KCl, 20 mM imidazole, 10% glycerol, 0.01% Triton X-100, 1 mM β-mercaptoethanol, and 0.1 mM PMSF. The protein mix was centrifuged to discard denatured proteins, and the supernatant was saved for Ni-NTA-agarose resin purification under native conditions. Then, 100 mL of protein mix was passed through 1 mL of settled ProBond Ni-NTA-agarose resin (Invitrogen, Waltham, MA, USA) previously equilibrated with the same dialysis buffer. Then, the resin was washed until no proteins were detected in the flow-through. Afterward, bound Fhl1 protein was recovered via elution with 200 mM imidazole in the dialysis buffer. The purified recombinant Fhl1 protein was dialyzed against 20 mM HEPES pH 7.9, 2.5 mM DTT, 50 mM KCl, 0.1 mM PMSF, 0.5 mM EDTA, and 20% v/v glycerol. The Fhl1 protein was analyzed with 10% SDS-PAGE, followed by silver staining.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}