1. Introduction
According to the European Society of Human Reproduction and Embryology (ESHRE) guidelines, RM is defined as the failure of two or more clinically recognized pregnancies before 20–24 weeks of gestation and includes embryonic and fetal losses [
1]. From an immunological perspective, since the embryo is a semi-allograft with paternal alloantigens, a successful pregnancy requires the maternal immune system to tolerate the semi-allogeneic fetus. The maternal–fetal interface, a dynamic site between the uterine mucosa and the extra-embryonic tissues of the developing conceptus, encompasses multiple cellular interactions in an environment rich in cytokines and hormones [
2]. Mounting evidence has demonstrated that disruption of maternal immune tolerance is one possible cause of RM. Previous studies have demonstrated that first-trimester human decidual leukocytes are primarily natural killer (NK) cells (~70%), macrophages (~20%), and T cells (10–20%), while dendritic cells (DCs), B cells, and natural killer T cells (NKT cells) are rare [
3]. A number of cytokines contribute to monocyte development into macrophages, which are known as uterine tissue-resident macrophages. There is a link between macrophages and embryo implantation, placental formation, embryonic development, and delivery [
4]. Together with our growing understanding of the roles and mechanisms of macrophages during pregnancy, current evidence suggests that macrophages are essential.
Mediator of IRF3 activation (MITA), also known as stimulator of interferon genes (STING), is a transmembrane protein that plays a critical role in virus-induced type I IFN signaling, as demonstrated by expression cloning. As we know, MITA was also found to localize to the outer membrane of mitochondria and to be associated with VISA, a mitochondrial protein that acts as an adaptor in virus-triggered signaling [
5]. Besides mediating the activation of IRF3 and NF-KB pathways and inducing the expression of IFN and proinflammatory cytokines, MITA also participates in other signal pathways and pathophysiological processes, such as autophagy, apoptosis, cell ageing, and the activation of inflammatory bodies [
6]. As a crucial mediator of inflammation during infection, cellular stress, and tissue injury, the cGAS-STING signaling pathway has emerged as a key pathway. Decout, A. et al. discuss the cGAS–STING signaling cascade and its association with degenerative, auto-inflammatory, and immune diseases [
7]. STING signaling could also be used to suppress covalently closed circular DNA (cccDNA) replication of hepatitis B virus (HBV) and alleviate liver fibrosis caused by HBV-induced macrophage inflammasome activation by activating autophagic flux in chronic HBV mice [
8]. MITA’s regulatory effect on macrophages in RM patients is still largely unknown due to the limited number of studies conducted.
Competition between endogenous RNAs (ceRNA) regulates the expression of each other by sharing miRNA response elements (MREs) [
9,
10]. In addition to being an alternative function for messenger RNAs, this hypothesis offers a unifying function for long non-coding RNAs, pseudogene transcripts, and circular RNAs [
11]. An intrinsic and exonic sequence are included within circular RNAs (circRNAs) formed by head-to-tail splicing of exons in a transcript. By forming a 3′,5′-phosphodiester bond between the 3′ end of an exon and the 5′ end of the upstream exon or its own, back-splicing junctions are created. circRNAs are stable transcripts found in both normal and malignant cells, and they may be encoded in thousands of human genes. Numerous studies have suggested that circRNAs may serve as promising disease markers due to their conservative properties [
12]. It has been demonstrated that circTP63 can compete with miR-873-3p, inhibiting miR-873-3p from downregulating FOXM1, which eventually facilitates cell cycle progression by increasing CENPA and CENPB [
13]. It has been suggested by Yao et al. [
14] that the epigenetic regulator PRMT5 inhibits CAMK2N1 transcription, which may be a potential therapeutic target for aggressive prostate cancer patients by acting on the circSPON2/miR-331-3p axis. It is not known whether circRNAs are responsible for RM in macrophages, and what their specific mechanisms are.
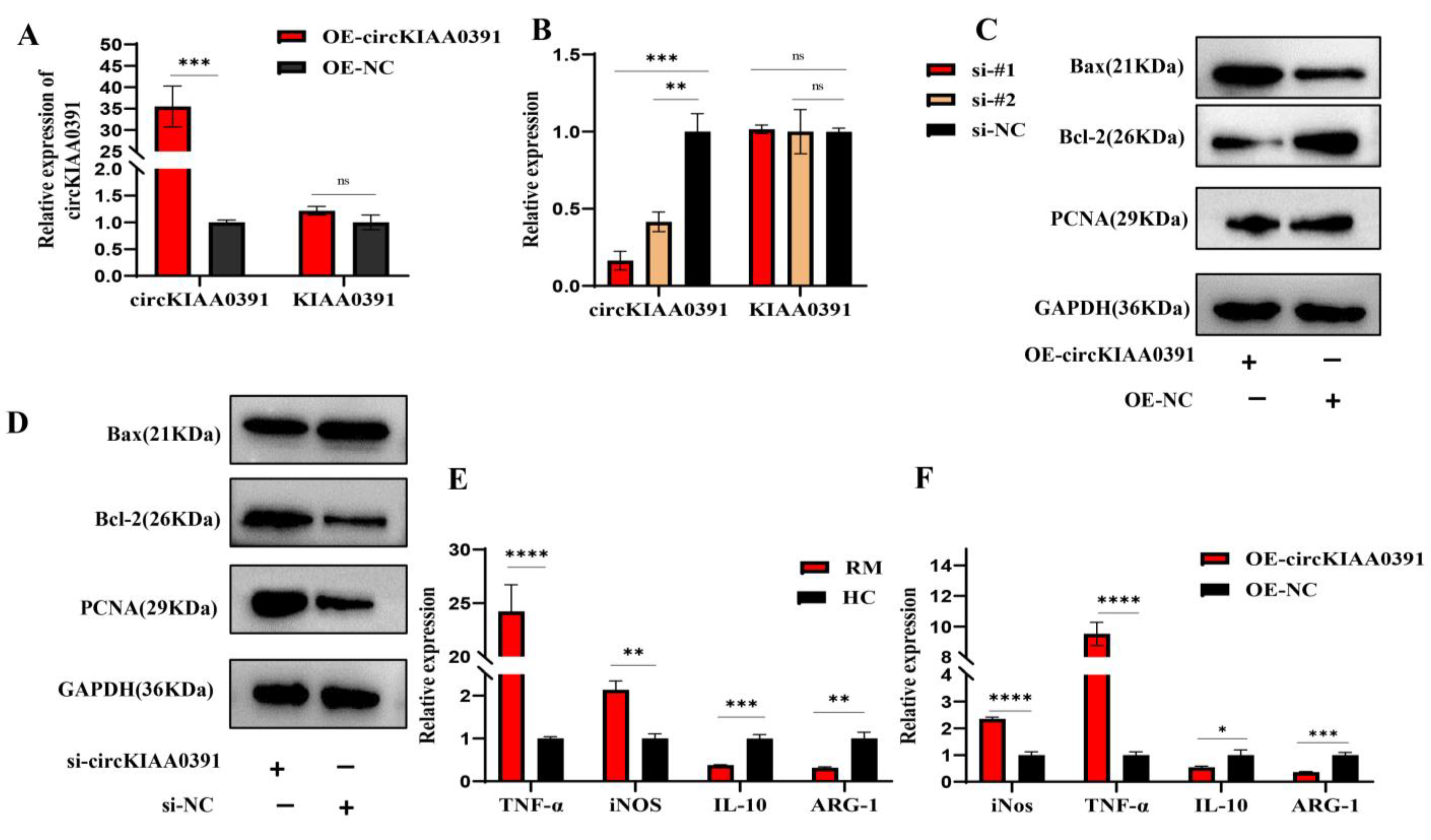
In the present study, we first found that MITA was significantly upregulated in RM patients and may be correlated to the pathology of RM. In order to further distinguish the regulatory network of MITA in macrophages, circRNA microarray profiling was conducted to construct the differential circRNA expression profiles in decidual macrophages and found that Hsa_circ_0003241 (also known as circKIAA0391) was significantly upregulated in RM patients. Further functional experiments demonstrated that circKIAA0391 could promote TDM cell apoptosis and proinflammatory polarization. Mechanistically, we further demonstrated that circKIAA0391 exerts its functions by absorbing the miR-512-5p/MITA axis in TDM cells, which may participate in the pathology of RM. Thus, circKIAA0391 might be a promising biomarker for the diagnosis and treatment of RM.
3. Discussion
RM is a common pregnancy complication with a multi-factorial etiology, representing a global public health challenge. As noted, MITA has been associated with infection as well as autoinflammatory, autoimmune, and degenerative diseases [
7,
24]. However, little is known about the expression of MITA in macrophages in RM. In this study, we first found that the ratio of M1/M2 in decidual tissues of RM patients was significantly higher than that in HC patients. We then found that the expression of MITA was upregulated in RM patients and M1 cells derived from TDM cells, as compared to the HC patients and M2 cells derived from TDM cells, respectively. Further results also demonstrated that MITA could directly interact with GSDMD or NLRP3 in TDM cells. These aforementioned results suggested that the high expression of MITA in decidual macrophages was closely related to the pathology of RM.
Recently, a body of evidence has demonstrated that MITA could act as an miRNA sponge to regulate the progression of many diseases. Chu, Q. et al. found that circRasGEF1B can bind to miR-21-3p directly and regulates MITA expression to enhance antiviral immunity in lower vertebrates [
25]. Yarbrough, M.L., et al. found that miR-576-3p is induced by IRF3 concomitantly with interferon and targets STING, MAVS, and TRAF3, which are critical factors for interferon [
26]. Furthermore, miR-181a could downregulate STING expression to suppress the activation of intrinsic interferons and may serve as a promising target for therapeutic exploitation in DNA oncoviruses [
27]. We hypothesized that the functions of MITA in macrophages might be regulated by non-coding RNAs (ncRNAs) in RM. In the present study, we first constructed differentially expressed circRNA profiles in the decidual macrophages of RM patients compared to the HC subjects. Then, we validated that circKIAA0391 was significantly upregulated in RM patients and M1 macrophages. In vivo studies also demonstrated that circKIAA0391 promotes macrophage apoptosis and proinflammatory polarization, indicating that circKIAA0391 in decidual macrophages might participate in the pathology of RM.
To our knowledge, exonic circRNAs are mainly located in the cytoplasm and could regulate gene expression in mammals at the transcriptional or posttranscriptional level by interacting with miRNAs [
28]. CircVAPA could activate the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway by modulating the miR-377-3p and miR-494-3p/insulin-like growth factor 1 receptor (IGF1R) axis to accelerate small cell lung cancer progression [
29]. Circ_0088194 could promote rheumatoid arthritis fibroblast-like synoviocytes (RA-FLSs) invasion and migration via the miR-766-3P/Matrix Metalloproteinase 2 (MMP2) axis [
30]. Furthermore, few studies have focused on the relationship between circRNA and miscarriage. Zhu et al. demonstrated that circPUM1 could impair the occurrence and development of RM by facilitating trophoblast cellular processes and protecting against inflammation via the miR-30a-5p/JUNB axis [
31]. Su et al. found that circCYP21A1 could play a role in RM by impairing the balance of cell proliferation and apoptosis by sponging miR-224, thereby regulating PRLR [
32]. However, the specific mechanism of circKIAA0391 in TDMs for RM still needs to be elucidated. In the present study, we identified that circKIAA0391 was mainly located in the cytoplasm by nucleic and cytoplasmic fractionation assay. Then, we further confirmed the direct binding relationship between miR-512-5p and circKIAA0391 by RIP, RNA pull-down, and dual-luciferase reporter assay.
Recently, evidence has been found that circRNA could act as ceRNA in TDM cells, influencing many diseases. Hsa_circ_0005567 overexpression promoted M2 macrophage polarization by binding to the miR-492/SOCS2 axis to reduce chondrocyte apoptosis, which could inhibit the progression of osteoarthritis [
33]. Zhang, C. et al. found that circPPM1F could act as a novel regulator of M1 macrophage activation through the circPPM1F-HuR-PPM1F-NF-κB axis [
34]. Mounting evidence indicates that miR-512-5p can sponge many circRNAs or LncRNAs to exert its degradation effect on target mRNAs, thus leading to the different phenotypes of cells. Circ_0000215 could exert oncogenic effects in nasopharyngeal carcinoma through the miR-512-5p/PIK3R1 axis [
35]. CircRPPH1 could act as an oncogene and regulate the progression of breast cancer via the circRPPH1-miR-512-5p-STAT1 axis [
36]. As we know, macrophages are essential for establishing and maintaining pregnancy by blood vessel remodeling, immune tolerance, immunomodulation of maternal decidual lymphocytes, and parturition initiation [
37]. Further studies have demonstrated that macrophages were skewed toward the M1 phenotype in spontaneous abortion [
38]. Moreover, the overexpression of circPPM1F could promote pancreatic islet injury by enhancing M1 macrophage activation, and circPPM1F may serve as a novel potential therapeutic target for type 1 diabetes in children [
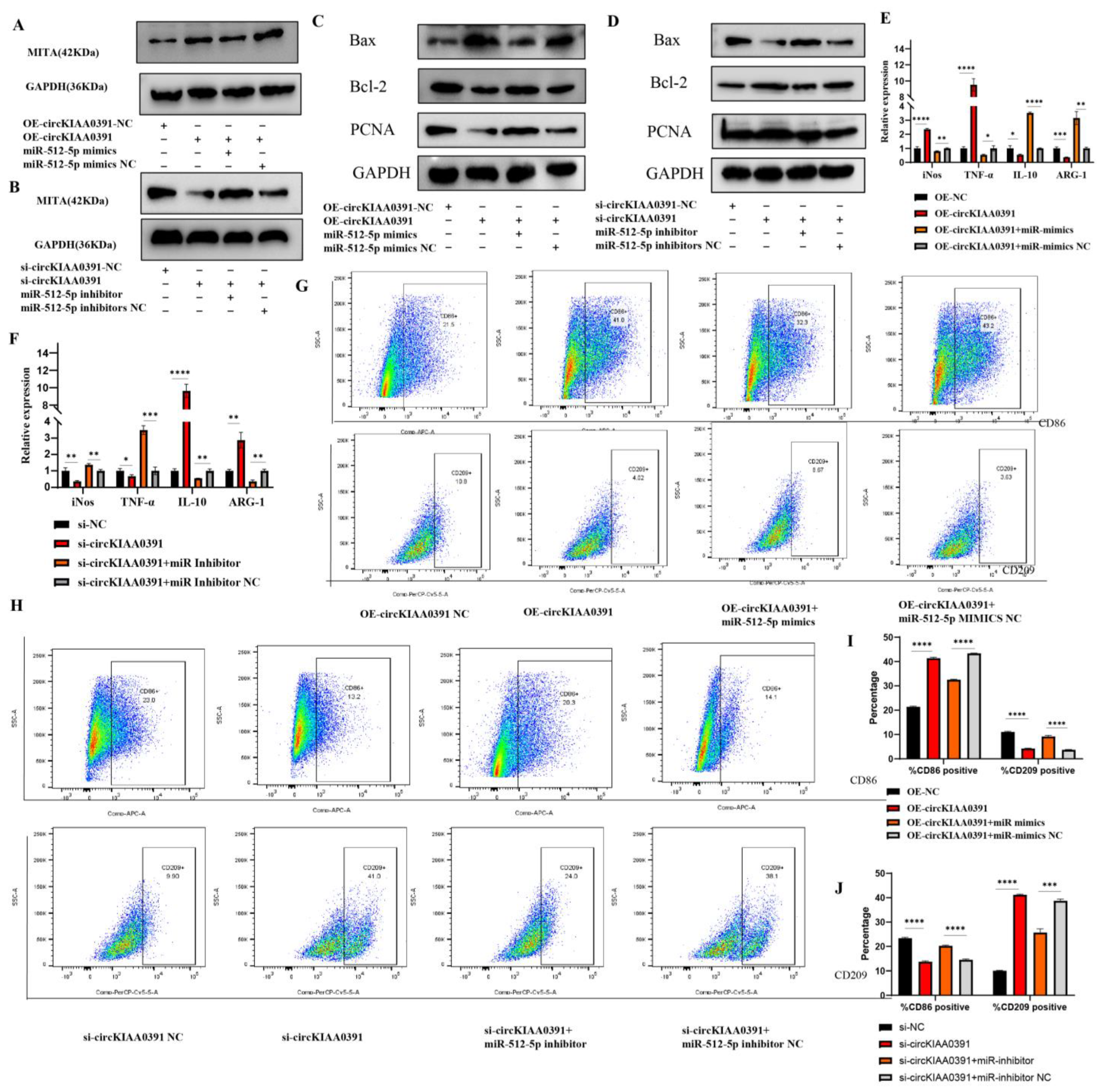
34]. To our knowledge, this was the first study to investigate the binding relationship between circKIAA0391/miR-512-5p and MITA. In this study, we ascertained the regulatory network of circKIAA0391/miR-512-5p on MITA by biotin-labeled miRNA pull-down assay. Mechanistically, the results also demonstrated that circKIAA0391 could regulate the expression of MITA by binding to miR-512-5p, thus mediating cell apoptosis and proinflammatory polarization in TDM cells. This study has provided a new perspective in research on the ceRNA regulatory network for MITA in TDM cells and a scientific basis for novel developments in the diagnosis and treatment of RM.
However, there are some shortcomings in the study to be acknowledged. Firstly, according to the nuclear and cytoplasm fractionation assay, about 40% of the expression of circKIAA0391 was distributed in the nuclear fraction. However, we only investigated the functions of circKIAA0391 in the cytoplasm in the present study; whether circKIAA0391 could regulate TDM cell apoptosis and macrophage proinflammatory polarization through other mechanisms, such as interacting with RNA-binding proteins and sponging trans-acting elements, requires further investigation. Furthermore, the mechanism of circKIAA0391/miR-512-5p/MITA on macrophage was validated in cell models. Further animal experiments and in vitro experiments are required to determine the mechanism.
4. Materials and Methods
4.1. Clinical Specimens and Ethics Statements
Uterine decidual tissues were collected from 25 healthy women and 25 patients with RM at Renmin Hospital of Wuhan University (Wuhan, China) between January 2018 and December 2021. When two or more pregnancies are lost sequentially before 20 weeks of gestation, it is referred to as RM. A group called HC included women with normal, early pregnancies who faced unwanted pregnancies and ended them by artificial abortion. Participants were excluded if they met any of the following conditions: 1. symptoms of endocrine or metabolic diseases, 2. abnormal karyotype analysis, and 3. uterine abnormality confirmed by pelvic examination or ultrasound. The basal characteristics of the included patients are listed in
Table 3. Decidual tissues were washed with iced phosphate buffer saline (PBS). Then, the tissues were ground to prepare the cell suspension. Monocytes were isolated from cell suspensions by density gradient centrifugation. After purification with anti-CD14-coated micro-beads (Thermofisher Scientific, Waltham, MA, USA), CD14-positive monocytes were sorted with a magnetic device. Informed consent was obtained from all patients before samples were collected, and all related procedures were approved by the internal review and ethics boards of the Renmin Hospital of Wuhan University.
4.2. RNA Extraction, cDNA Synthesis, Genome DNA Isolation, and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA extraction from cells/tissues was performed using Trizol reagent (TaKaRa Biotechnology, Dalian, China). Extracted total RNAs were measured by NanoDrop 2000 spectrophotometry (ThermoFisher Scientific, Waltham, MA, USA). For circRNA and mRNA, reverse transcriptions were performed using the PrimeScript RT Master Mix (TaKaRa Biotechnology, Dalian, China). For miRNA, reverse transcriptions were performed using the miRNA 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China) with specific sperm-loop primers. According to the manufacturer’s instructions, genome DNA was isolated using a QIAamp DNA Mini Kit (QIAGEN, Germantown, MD, USA). qRT-PCR analyses were performed using the ChamQ SYBR qPCR Master Mix (Vazyme, Nanjing, China). qRT-PCR was performed using the QuantStudio 5 Real-Time PCR system (ThermoFisher Scientific, Waltham, MA, USA), and relative gene expression was calculated by the 2
−∆∆CT method. The relative expressions of circRNA and mRNA were normalized to GAPDH, and U6 for miRNAs, respectively. The primers for circRNAs and mRNAs are listed in
Table 4, and the primers for miRNAs are listed in
Table 5.
4.3. Library Construction and Sequencing
Total RNAs were isolated from decidual macrophages in eight pairs of RM patients and healthy subjects. The quality of RNA samples was assessed and used to construct a library. Using a Ribo-Zero RNA Removal Kit (Illumina, San Diego, CA, USA), rRNA was removed from total RNA, then fragmented to approximately 200 bp with RNaseR. Following purification of the RNA fragments, cDNA first-strand and second-strand synthesis was performed, followed by adaptor ligation and low-cycle enrichment according to the instructions of NEBNext® UltraTM RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) and sequencing on the HiSeq 3000 using two 150 bp segments. We used EdgeR software (version 3.16.5) to normalize the data. To identify the differentially expressed circRNAs, |Log2Fold change| > 1 and p < 0.05 were selected as the cut-off criteria.
4.4. Functional Enrichment Analysis
The Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of the differentially expressed circRNAs were analyzed with the parental transcripts of these circRNAs. The GO categories were derived from Gene Ontology (
http://www.geneontology.org (accessed on 25 March 2021)) and comprised biological process (BP), molecular function (MF), and cellular component (CC). The KEGG database (
https://www.genome.jp/kegg (accessed on 25 March 2021)) is a widely used database storing information about genomes, biological pathways, diseases, and drugs. A
p-value of <0.05 was used as the threshold to indicate a significant difference.
4.5. Agarose Gel Electrophoresis and Sanger Sequencing
PCR products were subjected to the 0.8% agarose gel electrophoresis method. The back-splicing sequence of Hsa_circ_0003241 was verified through the circBase database (
http://www.circbase.org/ (accessed on 20 September 2021)). Amplification products of Hsa_circ_0003241 were confirmed by Sanger sequencing performed by Tsingke Biotech (Beijing, China).
4.6. Cell Line Culture and Macrophage Polarization
The human leukemia monocytic cells (THP-1) and the human embryonic kidney epithelial cell line (HEK-293T) were the kind gifts of Prof. Xi Zhou (State Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Academy of Science, Wuhan, China). The THP-1 cells were cultured in Roswell Park Memorial Institute (RMPI)-1640 medium (Thermo, Waltham, MA, USA) supplemented with 10% (volume fraction) fetal bovine serum (FBS, Gibco, Waltham, MA, USA) and 1% penicillin–streptomycin solution (P/S, Gibco, Waltham, MA, USA) at 37 °C and 5% CO2. THP-1-derived macrophage (TDM) cell differentiation was induced by stimulation with Phorbol-12-Myristate-13-Acetate (PMA, Sigma-Aldrich, Zwijndrecht, The Netherlands) for 24 h at a concentration of 100 ng/mL. After cell attachment, TDM cells were stimulated with LPS (20 ng/mL, Sigma-Aldrich, St. Louis, MO, USA) and IFN-γ (20 ng/mL, Invitrogen, Waltham, MA, USA) for M1 polarization or IL-4 (20 ng/mL, Invitrogen, Waltham, MA, USA) and recombinant human IL-13 (20 ng/mL, Sigma-Aldrich, St. Louis, MO, USA) for M2 polarization. Moreover, the HEK-293T cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Waltham, MA, USA) supplemented with 10% FBS and 1% P/S at 37 °C and 5% CO2.
4.7. RNaseR and Actinomycin D (Act-D) Treatment
To detect the stability of circRNA, RNaseR and Act-D treatment assays were performed. RNaseR, a member of the RNA superfamily of exoribonucleases, is a 3′-5′ exoribonuclease that releases 5′ nucleoside monophosphates. Briefly, total RNA samples extracted from TDM cells were incubated with or without 2 U/ug RNaseR (Geneseed Biotech, Guangzhou, China) at 37 °C for 15 min. Then, RNA was purified using the RNeasy MinElute Cleanup kit (Qiagen, Hilden, Germany). Furthermore, the Act-D treatment assay quantified their half-lives. The TDM cells were exposed to Act-D (Sigma-Aldrich, St. Louis, MO, USA) for 0, 4, 8, 12, and 24 h at a concentration of 2 µg/mL against new RNA synthesis. Then, the relative expression levels of RNAs were measured using the qRT-PCR assay.
4.8. Nuclear and Cytoplasmic Separation
Nuclear and cytoplasmic separation was performed following the manufacturer’s protocol. In brief, RNA was isolated from nuclear and cytoplasmic fractions using a Paris kit (Invitrogen, Waltham, MA, USA). Subsequently, the relative expression of circKIAA0391 was explored by qRT-PCR. GAPDH was the cytoplasmic control, while U6 was the nuclear control.
4.9. Lentivirus Package and Cell Infection
The circKIAA0391 overexpression plasmid (OE-circKIAA0391) was generated by inserting the full length of the circKIAA0391 sequence into a lentiviral vector (Tsingke, Wuhan, China). LiposofectamineTM 2000 (Invitrogen, Waltham, MA, USA) was used to co-transfect the overexpression plasmid with the psAX2 packaging plasmid and the pMD2.G envelope plasmid. A cellulose acetate filter (Millipore, Burlington, MA, USA) was used to filter the lentivirus supernatants collected 48 h post-transfection. Then, the THP-1 cells were infected by the recombinant lentivirus carrying circKIAA0391 overexpression plasmids or negative control plasmids for 48 h. The cells were subsequently selected using puromycin resistance for 1 week, and the surviving cells were regarded as stable circKIAA0391 overexpression cells or their matching control cells. Then, the relative expression of circKIAA0391 was examined by qRT-PCR.
4.10. Prediction of Target miRNA and miRNA Target Genes
4.11. Cell Transfection
siRNAs targeting the back-spliced sequence of circKIAA0391 were designed and synthesized by Ribobio Biotech (Guangzhou, China). To ensure the knockdown efficiency, two siRNA sequences were synthesized. Furthermore, miR-512-5p mimics (50 nM), miR-512-5p inhibitors (50 nM), and their matching negative controls were obtained from GenePharma Co. (Shanghai, China). Lipofectamine
TM 2000 reagent was used for cell transfections. After transfection, cells were collected for the subsequent assays. The detailed sequence of RNA oligos is listed in
Table 6.
4.12. Protein Extraction and Immunoblotting
Cell lysates were prepared with cell lysis buffer (ThermoFisher Scientific, Waltham, MA, USA). The total protein was quantified with the BCA Protein Assay Kit (Beyotime, Haimen, China) and denatured at 95 °C for 10 min. Next, 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate the protein, which was then transferred to polyvinylidene fluoride (PVDF) membranes. Then, incubation for 1 h at room temperature in Tris-buffered saline with Tween-20 (TBST) and 5% skim milk was performed to block the membrane. After that, the membrane was probed overnight at 4 °C using primary antibodies recognizing MITA (Abcam, Waltham, MA, USA, ab239074), PCNA (Abcam, Waltham, MA, USA, ab92552), BAX (1:1000, Abcam, Waltham, MA, USA, ab32503), Bcl-2 (Abcam, Waltham, MA, USA, ab182858), NLRP3 (Abcam, Waltham, MA, USA, ab263899), GSDMD (Abcam, Waltham, MA, USA, ab219800), cleaved N-terminal GSDMD (Abcam, Waltham, MA, USA, ab215203), caspase-1 (Abcam, Waltham, MA, USA, ab207802), HA tag (Abcam, Waltham, MA, USA, ab9110), DDDDK tag (Abcam, Waltham, MA, USA, ab205606), and GAPDH (1:2500, Abcam, Waltham, MA, USA, ab9485). After washing with TBST buffer three times, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat-anti-rabbit antibody (1:10,000, Abcam, Waltham, MA, USA) at room temperature for 1 h. Finally, after washing again with TBST solution five times, the bands were detected by ECL (Beyotime Biotechnology).
4.13. Dual-Luciferase Reporter Assay
The potential binding sites between miR-512-5p and circKIAA0391 were predicted using the TargetScan database. The wild-type and mutated sequences of circKIAA0391 (circKIAA0391-WT and circKIAA0391-Mut) were inserted in the pmiR-RB-ReportTM plasmid (Ribobio, Guangzhou, China). We transfected TDM cells with luciferase reporter plasmids and miR-512-5p mimics or the negative control (NC) for 24 h in 24-well plates. Following the manufacturer’s instructions, the dual-luciferase reporter assay system (Promega, Madison, WI, USA) was used to measure the luciferase activity. Renilla luciferase activity was used as a control for normalizing the results.
4.14. RNA Pull-Down Assay
Sense- and antisense-circKIAA0391 probes were synthesized and labeled with biotin by Ribobio Co. Ltd. (Guangzhou, China). The primers for biotin-labeled circKIAA0391 probes are listed in
Table 7. Biotinylated miR-512-5p and biotinylated NC probes were synthesized by GenePharma (Shanghai, China). The miRNA probes are listed in
Table 8. In brief, TDM cells were collected, and whole-cell lysates were extracted, followed by incubation with each probe at 4 °C for 4 h. Then, streptavidin-coupled agarose beads were added to the cell lysates for another 2 h. After washing, the biotin-coupled RNA complex was pulled down and extracted. Then, the abundances of circKIAA0391, miR-512-5p, and predicted target mRNAs were detected by qRT-PCR.
4.15. Immunoprecipitation (IP), Co-Immunoprecipitation (CO-IP), and RNA Immunoprecipitation
HEK-293T cells were transiently co-transfected for co-IPs with HA-MITA and Flag-GSDMD or HA-MITA and Flag-NLRP3. Forty-eight hours later, cells were harvested, and the protein amounts were determined using the BCA assay. Cell extracts were incubated for 12–16 h with the indicated antibodies, followed by additional incubation with protein A/G beads (Invitrogen, Waltham, MA, USA) for 2–4 h. For endogenous co-IP, antibodies were conjugated to resin using the CO-IP kit (Pierce, Appleton, WI, USA). The experiments were carried out in the presence of the proteasome inhibitor MG132 (20 μM). Input and eluted proteins from the pull-down and co-immunoprecipitation assays were separated by SDS-PAGE and probed with the corresponding antibodies in Western blot analyses. The RIP experiments were conducted using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Bedford, MA, USA), following the manufacturer’s instructions. In brief, TDM cells were transfected with miR-512-5p mimics or miR-NC. Cells were harvested after transfection for 48 h and lysed in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) with protease inhibitor cocktail (Invitrogen, Waltham, MA, USA) and RNasin® Plus Ribonuclease Inhibitor (Promega, Madison, WI, USA). Before IP, a positive control in the RIP assay, called “input”, was obtained from the cell lysate. Then, the remaining lysate was cultivated with magnetic beads, coupled with the AGO2 antibody (Abcam, Waltham, MA, USA, ab186733) or negative control IgG antibody (Proteintech, B900620, Wuhan, China), with rotation overnight at 4 °C. Finally, the RNA was purified from the RNA–protein complexes bound to the beads, and then the circKIAA0391 and miR-512-5p were determined using qRT-PCR.
4.16. Flow Cytometry (FCM)
For flow cytometry and immunofluorescence staining, CD206 was initially selected as a marker of M2, but our preliminary study found a low positive rate for CD206 in flow cytometry, leading us to hypothesize that CD206 would be required to break the membrane of the cells when performing the assay. Thus, the surface antigen CD209 was selected as a marker for M2 cells since another part of our study required multiple M2 cells for flow cytometric sorting. Collectively, to detect the polarization status of macrophages, we used qRT-PCR and flow cytometry. The mRNA level of iNOS and TNF-α and the surface marker of CD86 were used as the marker of M1. Furthermore, the mRNA level of ARG-1 and IL-10 and the surface marker of CD209 were used as the marker of M2. We still used CD206 as a marker for immunofluorescence since this experiment was performed at the tissue level. Using qRT-PCR and flow cytometry, we determined the polarization of macrophages and the combination of both data made the results more convincing.
Macrophage phenotypes were analyzed via the surface expression of specific surface markers. In the case of patients who underwent an abortion, the decidual tissue was collected and washed with PBS to remove any residual blood left behind. The tissue specimens were transferred to an ultra-clean bench at 4 °C to be processed. As a first step, the tissues were cut into small pieces with surgical scissors and ground to prepare a suspension of the cells. After centrifuging the cells and removing residual erythrocytes using an erythrocyte lysis solution, the lymphocytes were separated with a lymphocyte isolation solution. We harvested and washed the cells with ice-cold PBS and adjusted the cell concentration to 106 cells/mL. The cells were then immunolabeled with APC-CY7-conjugated Fixable Viability Stain780 (Becton, Dickinson and Company, Franklin Lakes, NJ, USA), APC-conjugated anti-human CD86 antibodies (Biolegend, San Diego, CA, USA) or Percp cy5.5-conjugated anti-human CD209 antibodies (Biolegend, San Diego, CA, USA). Then, the cells were incubated with the appropriate conjugated primary antibody for 30 min at 4 °C in the dark. The cells were centrifuged at 500 g/min and resuspended in PBS solution. Cells were then filtered through nylon mesh filters and washed with PBS. All of the data were acquired through flow cytometry (FACS Canto II; Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and processed using the FlowJo software 10.8.1 (Tree Star, Ashland, OR, USA).
4.17. Immunohistochemistry (IHC)
For the preparation of paraffin-embedded sections, fresh decidual tissues from RM and HC patients were fixed in 10% formalin for 24 h at room temperature, washed twice with PBS, and then stored in 70% ethanol solution at 4 °C until they were embedded in paraffin and sectioned at 4 μm. In each group, immunohistochemistry for MITA was performed. The paraffin-embedded sections were heated for 30 min at 65 °C, deparaffinized in xylene, and rehydrated through graded ethanol at room temperature. A 0.05 M Tris HCl buffer (pH 7.6) was used to prepare solutions and for washes between various steps. Incubations were performed in a humidified chamber. Sections were treated for 40 min at room temperature with 2% BSA and incubated overnight at 4 °C with primary antibodies. Horseradish peroxidase activity was visualized by treatment with H2O2 and 3,3′-diaminobenzidine for 5 min.
4.18. Multiplexed Immunofluorescence (mIF)
A Bond RX automated stainer (Leica Biosystems, Buffalo Grove, IL) was used to stain 4 mm thick formalin-fixed, paraffin-embedded decidual tissues. After incubating the cells for an hour in 5% BSA (0.1% Triton X-100), nonspecific binding was blocked. Anti-CD86 antibodies (Abcam, Waltham, MA, USA, ab239075), CD206 antibodies (Abcam, Waltham, MA, USA, ab64693), and MITA antibodies were incubated on the slides. Afterward, they were incubated with Cy3, fluorescein isothiocyanate (FITC), or Cy5. The nuclei of the cells were stained using DAPI. The images were observed by using a confocal microscope.
4.19. Statistical Analysis
Analysis of the collection and statistical data were processed with the SPSS software version 22.0 (SPSS, Chicago, IL, USA) using Student’s t-test. All of the experiments were repeated at least three times for each group. Correlations were performed by Pearson correlation analysis. Data were expressed as the means ± standard deviation and were plotted using the GraphPad Prism program version 9.4.0 (GraphPad Prism8; GraphPad, San Diego CA, USA). Two-sided p < 0.05 was considered to represent a statistically significant difference.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}