
Characterization of Unidirectional Replication Forks in the Mouse Genome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
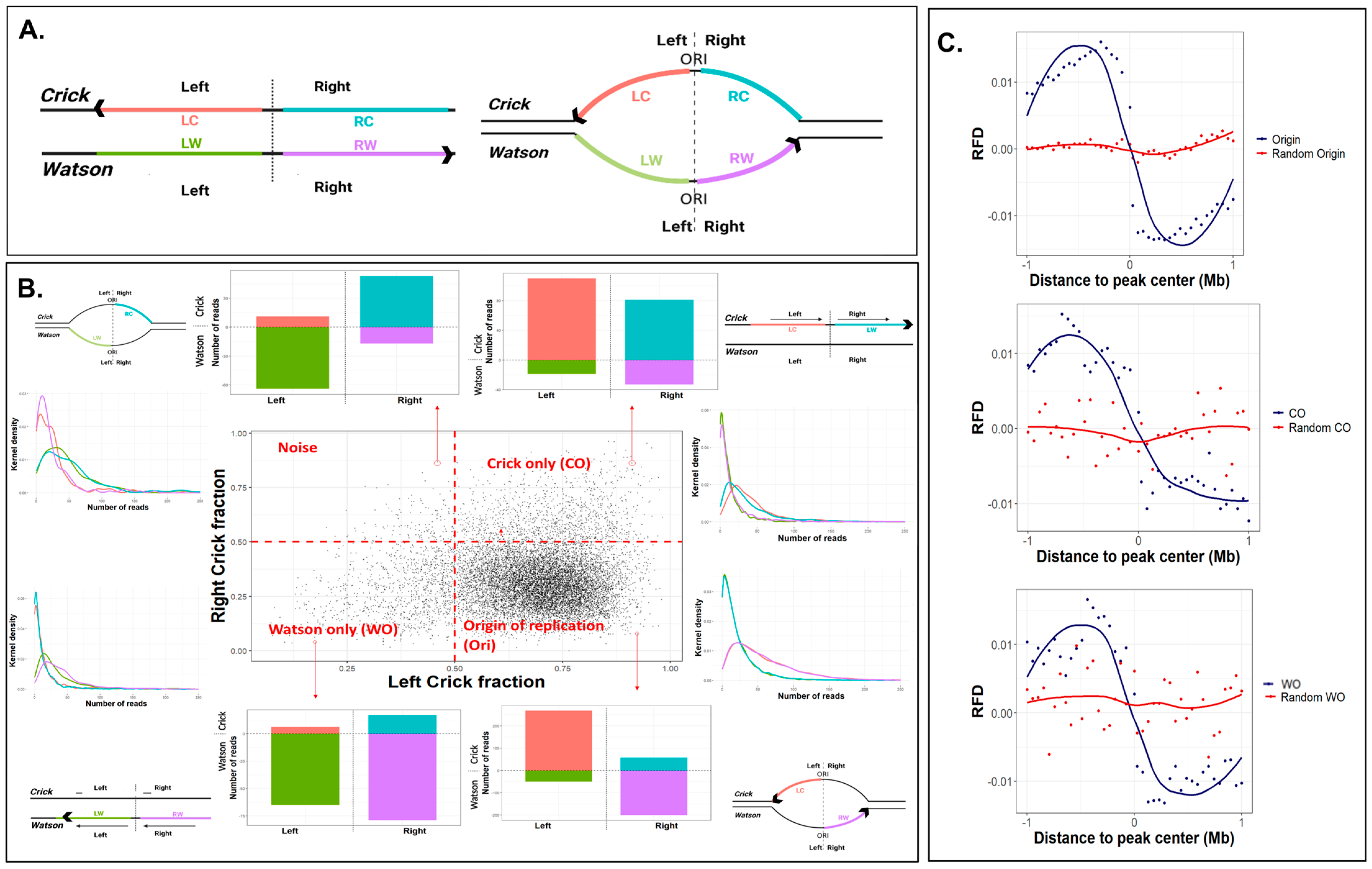
2.1. Single-Stranded Ori-SSDS Peaks Are Origin of Replication
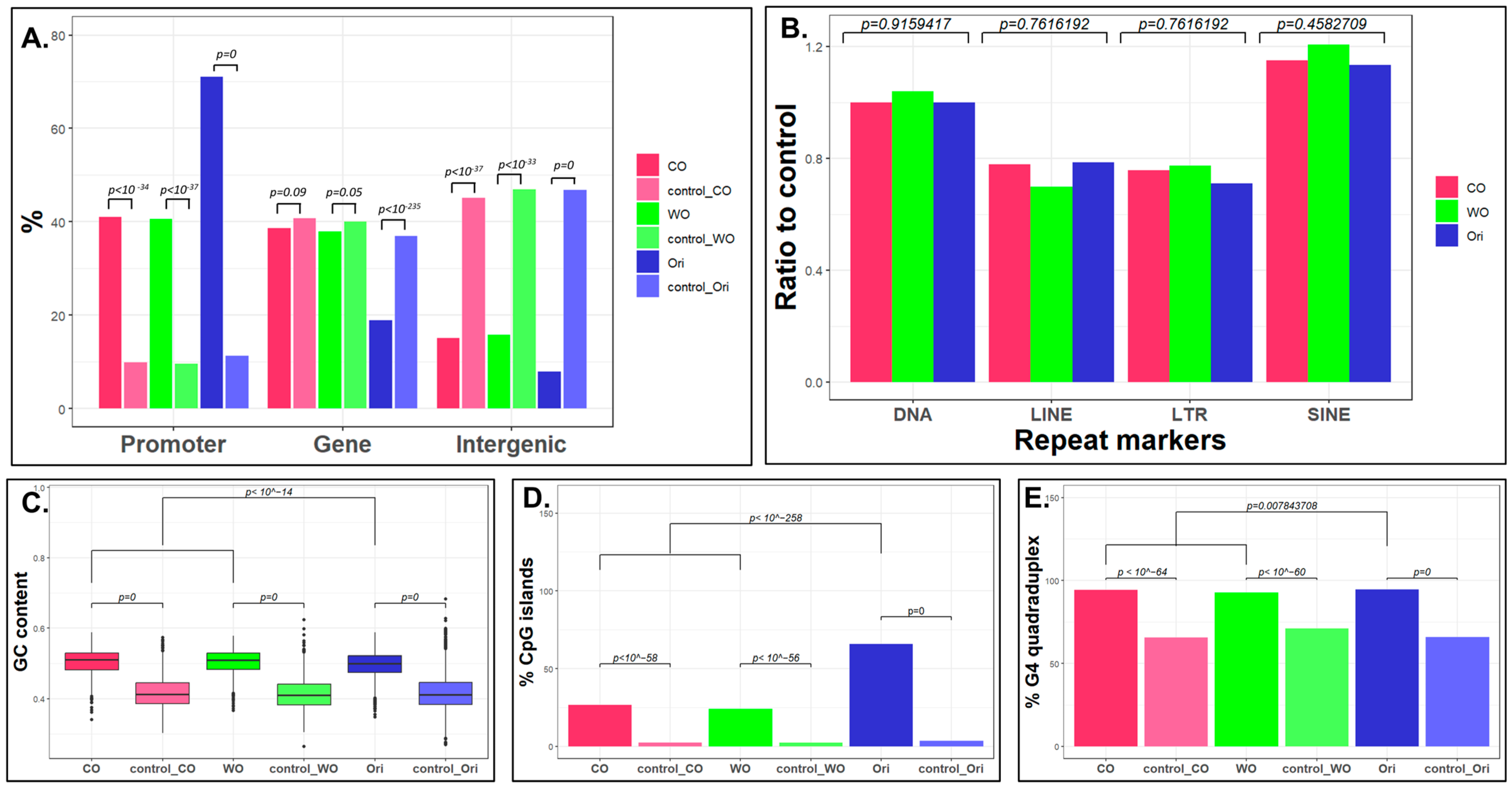
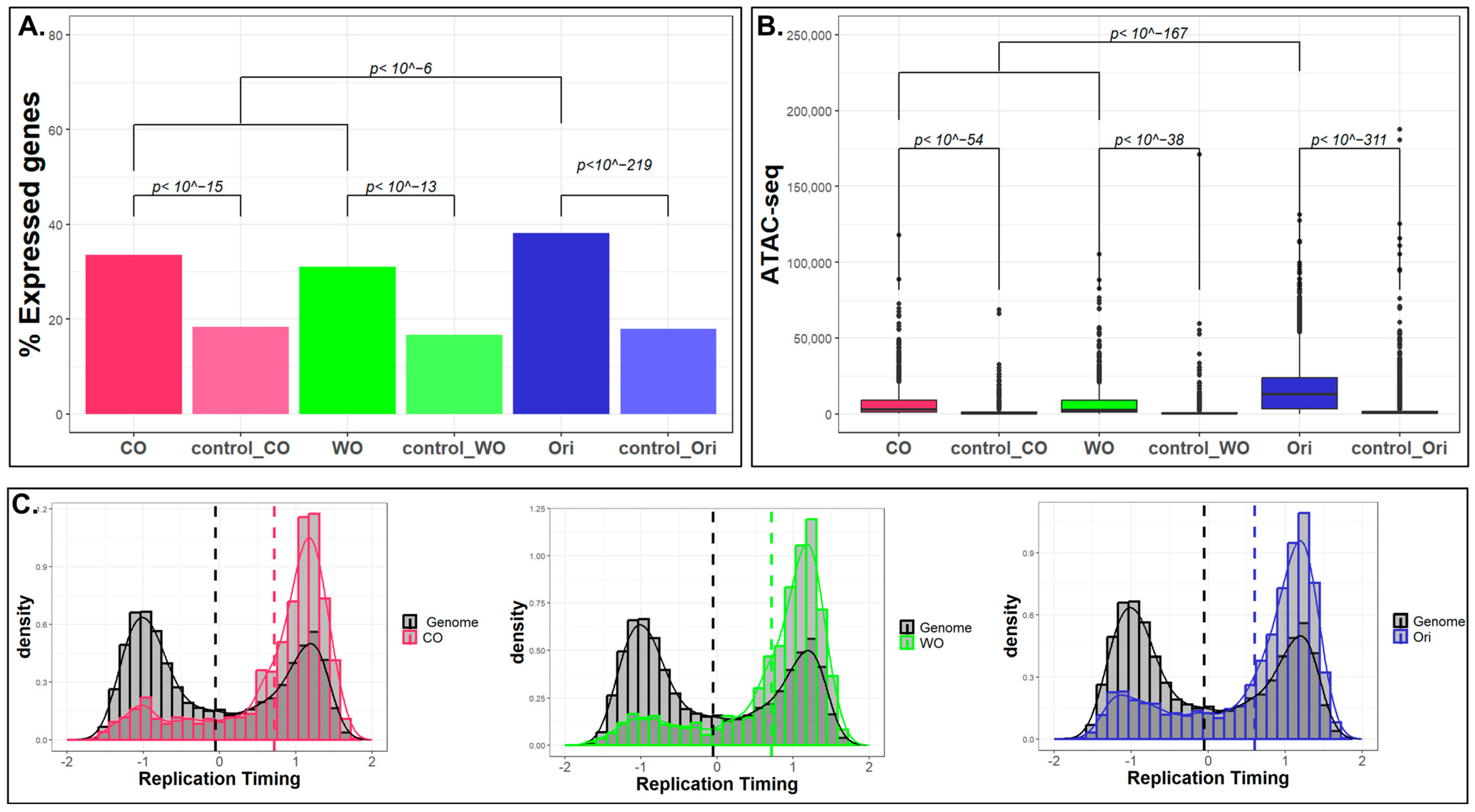
2.2. Characterization of the Unidirectional Origins
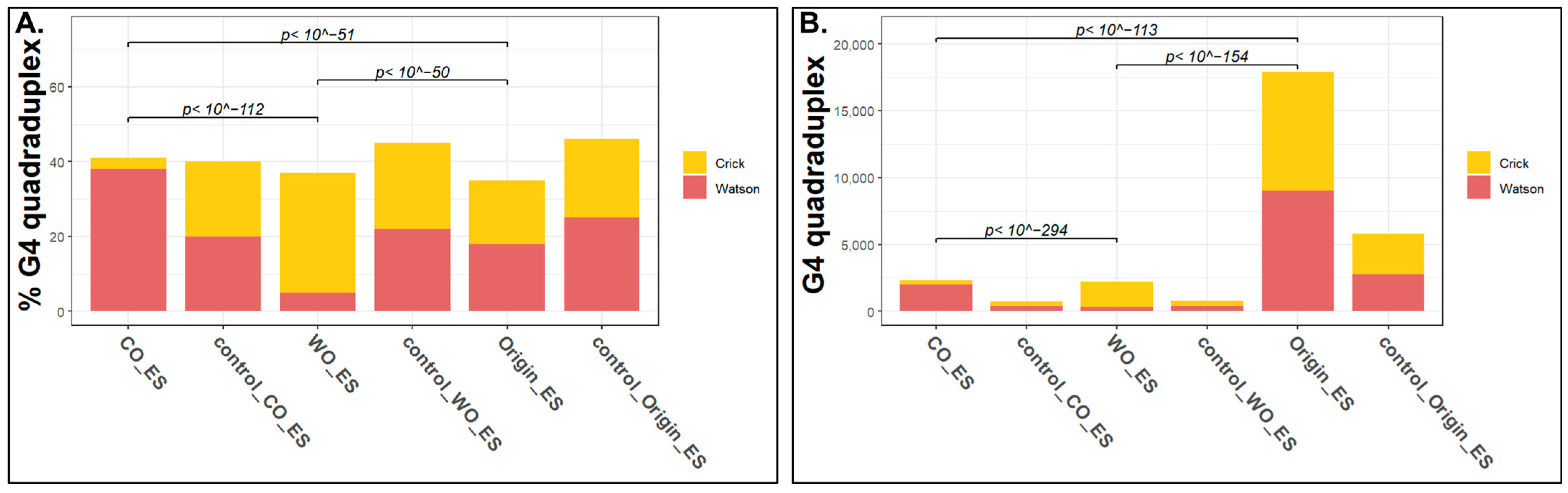
2.3. What Is Unique in Unidirectional Origins?
3. Discussion
4. Conclusions
5. Methods
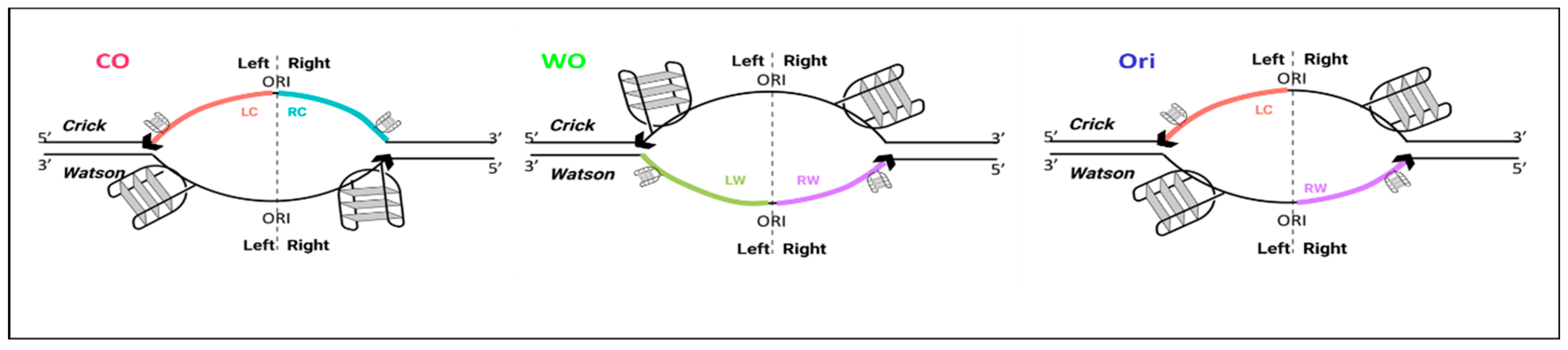
5.1. Definition of CO, WO, and Origin Peaks
5.2. Randomize Regions
5.3. Okazaki Sequencing Data Processing
5.4. Replication Fork Directionally
5.5. Genomics Feature Analysis
5.6. Epigenomics Feature Analyses
5.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mechali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Mechali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Ekundayo, B.; Bleichert, F. Origins of DNA replication. PLoS Genet. 2019, 15, e1008320. [Google Scholar] [CrossRef] [PubMed]
- Jaksik, R.; Wheeler, D.; Kimmel, M. Detection and characterization of constitutive replication origins defined by DNA polymerase epsilon. BMC Biol. 2023, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.; Johnson, E.M. Mapping initiation sites of DNA replication in vivo using polymerase chain reaction amplification of nascent strand segments. Nucleic Acids Res. 1989, 17, 7693–7705. [Google Scholar] [CrossRef] [PubMed]
- Pratto, F.; Brick, K.; Cheng, G.; Lam, K.-W.G.; Cloutier, J.M.; Dahiya, D.; Wellard, S.R.; Jordan, P.W.; Camerini-Otero, R.D. Meiotic recombination mirrors patterns of germline replication in mice and humans. Cell 2021, 184, 4251–4267.e20. [Google Scholar] [CrossRef] [PubMed]
- Mourón, S.; Acebes, S.R.; I Martínez-Jiménez, M.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Mendez, J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef]
- Bailey, L.J.; Bianchi, J.; Doherty, A.J. PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res. 2019, 47, 4026–4038. [Google Scholar] [CrossRef]
- Hizume, K.; Araki, H. Replication fork pausing at protein barriers on chromosomes. FEBS Lett. 2019, 593, 1449–1458. [Google Scholar] [CrossRef]
- Neylon, C.; Kralicek, A.; Hill, T.; Dixon, N.E. Replication termination in Escherichia coli: Structure and antihelicase activity of the Tus-Ter complex. Microbiol. Mol. Biol. Rev. 2005, 69, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Linskens, M.H.; Huberman, J.A. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol. Cell Biol. 1988, 8, 4927–4935. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, R.; Michel, B.; Gangloff, S. Replication fork pausing and recombination or “gimme a break”. Genes Dev. 2000, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shyian, M.; Shore, D. Approaching Protein Barriers: Emerging Mechanisms of Replication Pausing in Eukaryotes. Front. Cell Dev. Biol. 2021, 9, 672510. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.K.; Sale, J.E. Replication of G Quadruplex DNA. Genes 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef]
- Lee, W.T.C.; Yin, Y.; Morten, M.J.; Tonzi, P.; Gwo, P.P.; Odermatt, D.C.; Modesti, M.; Cantor, S.B.; Gari, K.; Huang, T.T.; et al. Single-molecule imaging reveals replication fork coupled formation of G-quadruplex structures hinders local replication stress signaling. Nat. Commun. 2021, 12, 2525. [Google Scholar] [CrossRef]
- Sato, K.; Martin-Pintado, N.; Post, H.; Altelaar, M.; Knipscheer, P. Multistep mechanism of G-quadruplex resolution during DNA replication. Sci. Adv. 2021, 7, eabf8653. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Wu, Y. An emerging picture of FANCJ’s role in G4 resolution to facilitate DNA replication. NAR Cancer 2021, 3, zcab034. [Google Scholar] [CrossRef]
- Paeschke, K.; Burkovics, P. Mgs1 function at G-quadruplex structures during DNA replication. Curr. Genet. 2021, 67, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Freudenreich, C.H. A Timeless Tale: G4 structure recognition by the fork protection complex triggers unwinding by DDX11 helicase. EMBO J. 2020, 39, e106305. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Dalby, M.; Wenger, A.; Stromme, C.B.; Strandsby, A.; Andersson, R.; Groth, A. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 2018, 361, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Puy, A.; Rialle, S.; Kaplan, N.; Segal, E.; Méchali, M. New insights into replication origin characteristics in metazoans. Cell Cycle 2012, 11, 658–667. [Google Scholar] [CrossRef]
- Marsico, G.; Chambers, V.S.; Sahakyan, A.B.; McCauley, P.; Boutell, J.M.; Di Antonio, M.; Balasubramanian, S. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 2019, 47, 3862–3874. [Google Scholar] [CrossRef]
- Gajewska, K.A.; Ramialison, M.; Wagstaff, K.; Jans, D.A. Transcriptomic profile dataset of embryonic stem cells (Wild-type and IPO13-Knock Out) with and without oxidative stress. Data Brief 2022, 42, 108099. [Google Scholar] [CrossRef]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef]
- Zhao, P.A.; Sasaki, T.; Gilbert, D.M. High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol. 2020, 21, 76. [Google Scholar] [CrossRef]
- Yehuda, Y.; Blumenfeld, B.; Mayorek, N.; Makedonski, K.; Vardi, O.; Cohen-Daniel, L.; Mansour, Y.; Baror-Sebban, S.; Masika, H.; Farago, M.; et al. Germline DNA replication timing shapes mammalian genome composition. Nucleic Acids Res. 2018, 46, 8299–8310. [Google Scholar] [CrossRef]
- Schiavone, D.; Jozwiakowski, S.K.; Romanello, M.; Guilbaud, G.; Guilliam, T.A.; Bailey, L.J.; Sale, J.E.; Doherty, A.J. PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol. Cell 2015, 61, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.; Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.-P.; Foiani, M.; Nicolas, A. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011, 30, 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Murat, P.; Phillips, L.; Patel, K.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2011, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, C.; Guilbaud, G.; Schiavone, D.; Sale, J.E. Nucleotide Pool Depletion Induces G-Quadruplex-Dependent Perturbation of Gene Expression. Cell Rep. 2015, 13, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Shao, R.; Yung, P.K.; Elsasser, S.J. Genome-wide mapping of G-quadruplex structures with CUT&Tag. Nucleic Acids Res. 2021, 50, e13. [Google Scholar] [CrossRef]
- Labib, K.; Hodgson, B. Replication fork barriers: Pausing for a break or stalling for time? EMBO Rep. 2007, 8, 346–353. [Google Scholar] [CrossRef]
- Beuzer, P.; Quivy, J.; Almouzni, G. Establishment of a replication fork barrier following induction of DNA binding in mammalian cells. Cell Cycle 2014, 13, 1607–1616. [Google Scholar] [CrossRef]
- Willis, N.A.; Panday, A.; Duffey, E.; Scully, R. Rad51 recruitment and exclusion of non-homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS Genet. 2018, 14, e1007486. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zerbib, A.; Simon, I. Characterization of Unidirectional Replication Forks in the Mouse Genome. Int. J. Mol. Sci. 2023, 24, 9611. https://doi.org/10.3390/ijms24119611
Zerbib A, Simon I. Characterization of Unidirectional Replication Forks in the Mouse Genome. International Journal of Molecular Sciences. 2023; 24(11):9611. https://doi.org/10.3390/ijms24119611
Chicago/Turabian StyleZerbib, Avital, and Itamar Simon. 2023. "Characterization of Unidirectional Replication Forks in the Mouse Genome" International Journal of Molecular Sciences 24, no. 11: 9611. https://doi.org/10.3390/ijms24119611
APA StyleZerbib, A., & Simon, I. (2023). Characterization of Unidirectional Replication Forks in the Mouse Genome. International Journal of Molecular Sciences, 24(11), 9611. https://doi.org/10.3390/ijms24119611