
Type 2 Diabetes Mellitus, Non-Alcoholic Fatty Liver Disease, and Metabolic Repercussions: The Vicious Cycle and Its Interplay with Inflammation
 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Focal Question
2.2. Language
2.3. Databases
2.4. Study Extraction
2.5. Data Extraction
2.6. Quality Assessment
3. NAFLD and DM2 Coexistence
4. Inflammatory Mechanisms Underlying NAFLD and DM2 Pathogenesis
4.1. Inflammation as a Pathway of NAFLD Progression to NASH
4.2. Inflammation as a Factor in Insulin Resistance
4.3. Inflammation in the Pathogenesis of Type 2 Diabetes Mellitus
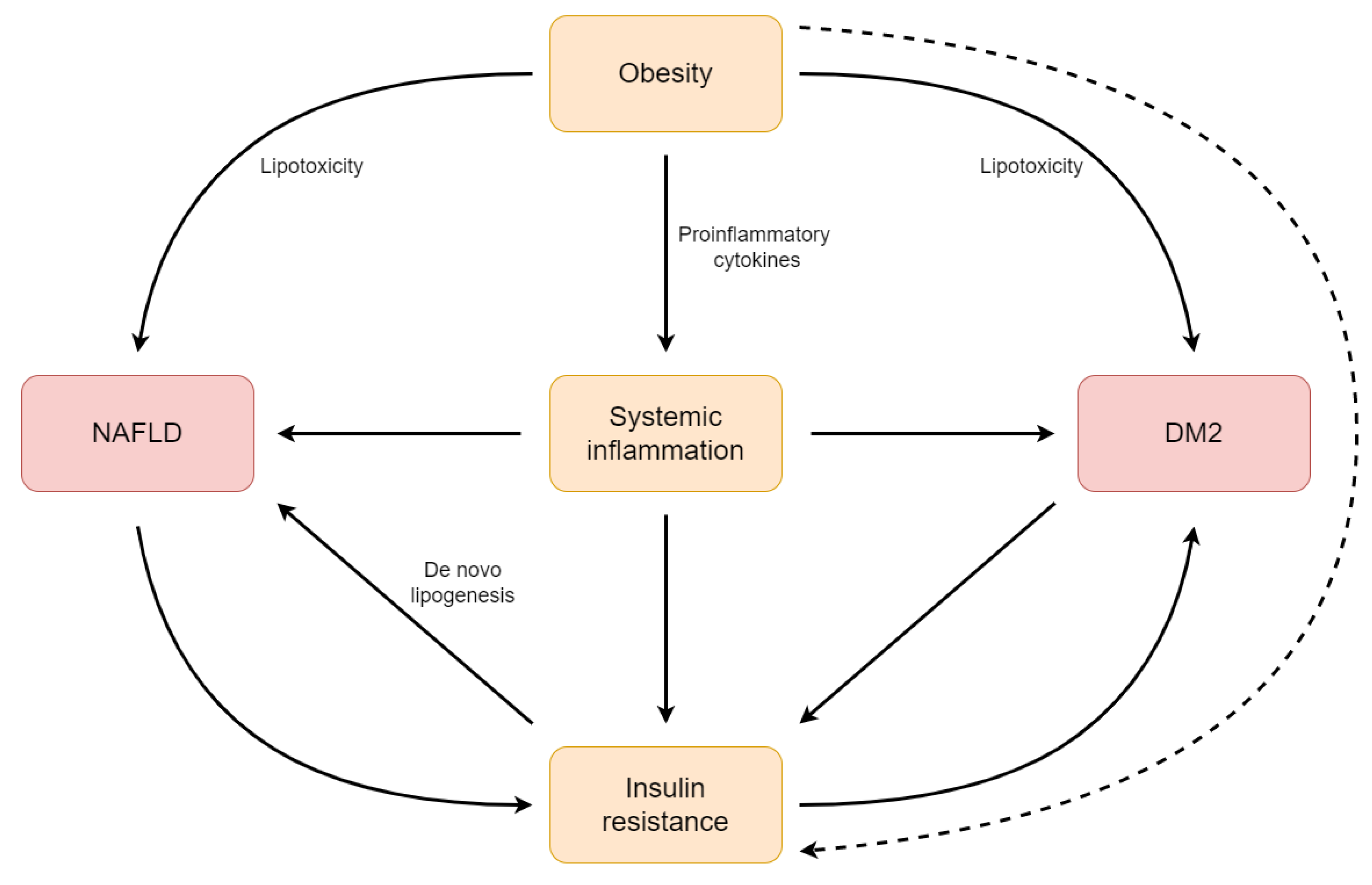
5. Inflammation as a Link between NAFLD and DM2
6. The Outlook of Anti-Inflammatory Treatment in NAFLD and DM2
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calabrese, F.M.; Disciglio, V.; Franco, I.; Sorino, P.; Bonfiglio, C.; Bianco, A.; Campanella, A.; Lippolis, T.; Pesole, P.L.; Polignano, M.; et al. A Low Glycemic Index Mediterranean Diet Combined with Aerobic Physical Activity Rearranges the Gut Microbiota Signature in NAFLD Patients. Nutrients 2022, 14, 1773. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The Prevalence and Incidence of NAFLD Worldwide: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Tao, G.; Zhang, G.; Chen, W.; Yang, C.; Xue, Y.; Song, G.; Qin, S. A Randomized, Placebo-controlled Clinical Trial of Hydrogen/Oxygen Inhalation for Non-alcoholic Fatty Liver Disease. J. Cell. Mol. Med. 2022, 26, 4113–4123. [Google Scholar] [CrossRef]
- Loomba, R.; Morgan, E.; Watts, L.; Xia, S.; Hannan, L.A.; Geary, R.S.; Baker, B.F.; Bhanot, S. Novel Antisense Inhibition of Diacylglycerol O-Acyltransferase 2 for Treatment of Non-Alcoholic Fatty Liver Disease: A Multicentre, Double-Blind, Randomised, Placebo-Controlled Phase 2 Trial. Lancet Gastroenterol. Hepatol. 2020, 5, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Kedarisetty, C.K.; Bhardwaj, A.; Kumar, G.; Rastogi, A.; Bihari, C.; Kumar, M.; Sarin, S.K. Efficacy of Combining Pentoxiphylline and Vitamin E versus Vitamin E Alone in Non-Alcoholic Steatohepatitis—A Randomized Pilot Study. Indian J. Gastroenterol. 2021, 40, 41–49. [Google Scholar] [CrossRef]
- Nakao, Y.; Amrollahi, P.; Parthasarathy, G.; Mauer, A.S.; Sehrawat, T.S.; Vanderboom, P.; Nair, K.S.; Nakao, K.; Allen, A.M.; Hu, T.Y.; et al. Circulating Extracellular Vesicles Are a Biomarker for NAFLD Resolution and Response to Weight Loss Surgery. Nanomed. Nanotechnol. Biol. Med. 2021, 36, 102430. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Chen, Q.; Wu, L.; He, W.; Tu, D.; Wang, S.; Chen, Y.; Liu, S.; Xie, Z.; et al. Disulfiram Ameliorates Nonalcoholic Steatohepatitis by Modulating the Gut Microbiota and Bile Acid Metabolism. Nat. Commun. 2022, 13, 6862. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Deng, Z.; Luo, W.; He, X.; Chen, Y. Effect of Fecal Microbiota Transplantation on Non-Alcoholic Fatty Liver Disease: A Randomized Clinical Trial. Front. Cell. Infect. Microbiol. 2022, 12, 759306. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Marchesini, G.; Petroni, M.L.; Cortez-Pinto, H. Adipose Tissue-Associated Cancer Risk: Is It the Fat around the Liver, or the Fat inside the Liver? J. Hepatol. 2019, 71, 1073–1075. [Google Scholar] [CrossRef] [Green Version]
- Le, M.H.; Yeo, Y.H.; Li, X.; Li, J.; Zou, B.; Wu, Y.; Ye, Q.; Huang, D.Q.; Zhao, C.; Zhang, J.; et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 2809–2817.e28. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, T.; Pantic, I.; Dragasevic, S.; Lugonja, S.; Dumic, I.; Rajilic-Stojanovic, M. The Interrelationship Among Non-Alcoholic Fatty Liver Disease, Colonic Diverticulosis and Metabolic Syndrome. JGLD 2021, 30, 1–9. [Google Scholar] [CrossRef]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diabetes Rep. 2021, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.-U.; Cusi, K. Non-Alcoholic Fatty Liver Disease: Causes, Diagnosis, Cardiometabolic Consequences, and Treatment Strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD as a Driver of Chronic Kidney Disease. J. Hepatol. 2020, 72, 785–801. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Schattenberg, J.M.; Tilg, H.; Byrne, C.D.; Targher, G. Non-Alcoholic Fatty Liver Disease and Risk of Incident Chronic Kidney Disease: An Updated Meta-Analysis. Gut 2022, 71, 156–162. [Google Scholar] [CrossRef]
- Nadinskaia, M.; Maevskaya, M.; Ivashkin, V.; Kodzoeva, K.; Pirogova, I.; Chesnokov, E.; Nersesov, A.; Kaibullayeva, J.; Konysbekova, A.; Raissova, A.; et al. Ursodeoxycholic Acid as a Means of Preventing Atherosclerosis, Steatosis and Liver Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2021, 27, 959–975. [Google Scholar] [CrossRef]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Eslam, M.; Kawaguchi, T.; Tsutsumi, T.; Nakano, D.; Yoshinaga, S.; Takahashi, H.; Anzai, K.; George, J.; Torimura, T. MAFLD Identifies Patients with Significant Hepatic Fibrosis Better than NAFLD. Liver Int. 2020, 40, 3018–3030. [Google Scholar] [CrossRef] [PubMed]
- Gofton, C.; Upendran, Y.; Zheng, M.-H.; George, J. MAFLD: What Is Different from NAFLD? Clin. Mol. Hepatol. 2023, 29, S17–S31. [Google Scholar] [CrossRef]
- Kharroubi, A.T.; Darwish, H.M. Diabetes Mellitus: The Epidemic of the Century. World J. Diabetes 2015, 6, 850–867. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes—2023. Diabetes Care 2022, 46, S19–S40. [Google Scholar] [CrossRef]
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic Complications of Diabetes Mellitus: A Mini Review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Ligthart, S.; Vaez, A.; Võsa, U.; Stathopoulou, M.G.; de Vries, P.S.; Prins, B.P.; Van der Most, P.J.; Tanaka, T.; Naderi, E.; Rose, L.M.; et al. Genome Analyses of >200,000 Individuals Identify 58 Loci for Chronic Inflammation and Highlight Pathways That Link Inflammation and Complex Disorders. Am. J. Hum. Genet. 2018, 103, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as Immune Regulators in Infection, Inflammation and Cancer. Nat. Rev. Immunol. 2021, 21, 704–717. [Google Scholar] [CrossRef]
- Ehses, J.A.; Ellingsgaard, H.; Böni-Schnetzler, M.; Donath, M.Y. Pancreatic Islet Inflammation in Type 2 Diabetes: From α and β Cell Compensation to Dysfunction. Arch. Physiol. Biochem. 2009, 115, 240–247. [Google Scholar] [CrossRef]
- Fisk, H.L.; Childs, C.E.; Miles, E.A.; Ayres, R.; Noakes, P.S.; Paras-Chavez, C.; Kuda, O.; Kopecký, J.; Antoun, E.; Lillycrop, K.A.; et al. Modification of Subcutaneous White Adipose Tissue Inflammation by Omega-3 Fatty Acids Is Limited in Human Obesity-a Double Blind, Randomised Clinical Trial. eBioMedicine 2022, 77, 103909. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Liani, R.; Tripaldi, R.; Di Castelnuovo, A.; Guagnano, M.; Tartaro, A.; Bonadonna, R.; Federico, V.; Cipollone, F.; Consoli, A.; et al. Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss. Nutrients 2018, 10, 1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, D.; Welch, B.S.; Rehman, A. Pathophysiology of Obesity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lima, M.M.O.; Pareja, J.C.; Alegre, S.M.; Geloneze, S.R.; Kahn, S.E.; Astiarraga, B.D.; Chaim, É.A.; Baracat, J.; Geloneze, B. Visceral Fat Resection in Humans: Effect on Insulin Sensitivity, Beta-Cell Function, Adipokines, and Inflammatory Markers: Visceral Fat Resection in Humans. Obesity 2013, 21, E182–E189. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.F.; Pantoja, J.P.; Velázquez-Fernández, D.; Cabiedes, J.; Aguilar-Salinas, C.; García-García, E.; Rivas, A.; Villeda, C.; Hernández-Ramírez, D.F.; Dávila, A.; et al. Potential Additional Effect of Omentectomy on Metabolic Syndrome, Acute-Phase Reactants, and Inflammatory Mediators in Grade III Obese Patients Undergoing Laparoscopic Roux-En-Y Gastric Bypass. Diabetes Care 2010, 33, 1413–1418. [Google Scholar] [CrossRef] [Green Version]
- Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. [Google Scholar] [CrossRef]
- Pervez, M.A.; Khan, D.A.; Mirza, S.A.; Slehria, A.U.R.; Nisar, U.; Aamir, M. Comparison of Delta-Tocotrienol and Alpha-Tocopherol Effects on Hepatic Steatosis and Inflammatory Biomarkers in Patients with Non-Alcoholic Fatty Liver Disease: A Randomized Double-Blind Active-Controlled Trial. Complement. Ther. Med. 2022, 70, 102866. [Google Scholar] [CrossRef]
- Cheng, R.; Wang, L.; Le, S.; Yang, Y.; Zhao, C.; Zhang, X.; Yang, X.; Xu, T.; Xu, L.; Wiklund, P.; et al. A Randomized Controlled Trial for Response of Microbiome Network to Exercise and Diet Intervention in Patients with Nonalcoholic Fatty Liver Disease. Nat. Commun. 2022, 13, 2555. [Google Scholar] [CrossRef]
- Climax, J.; Newsome, P.N.; Hamza, M.; Weissbach, M.; Coughlan, D.; Sattar, N.; McGuire, D.K.; Bhatt, D.L. Effects of Epeleuton, a Novel Synthetic Second-Generation N-3 Fatty Acid, on Non-Alcoholic Fatty Liver Disease, Triglycerides, Glycemic Control, and Cardiometabolic and Inflammatory Markers. JAHA 2020, 9, e016334. [Google Scholar] [CrossRef]
- Musazadeh, V.; Dehghan, P.; Saleh-Ghadimi, S.; Abbasalizad Farhangi, M. Omega 3-rich Camelina Sativa Oil in the Context of a Weight Loss Program Improves Glucose Homeostasis, Inflammation and Oxidative Stress in Patients with NAFLD: A Randomised Placebo-controlled Clinical Trial. Int. J. Clin. Pract. 2021, 75, e14744. [Google Scholar] [CrossRef]
- Mosca, A.; Crudele, A.; Smeriglio, A.; Braghini, M.R.; Panera, N.; Comparcola, D.; Alterio, A.; Sartorelli, M.R.; Tozzi, G.; Raponi, M.; et al. Antioxidant Activity of Hydroxytyrosol and Vitamin E Reduces Systemic Inflammation in Children with Paediatric NAFLD. Dig. Liver Dis. 2021, 53, 1154–1158. [Google Scholar] [CrossRef]
- Alkhouri, N.; Berk, M.; Yerian, L.; Lopez, R.; Chung, Y.-M.; Zhang, R.; McIntyre, T.M.; Feldstein, A.E.; Hazen, S.L. OxNASH Score Correlates with Histologic Features and Severity of Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2014, 59, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Kosmalski, M.; Ziółkowska, S.; Czarny, P.; Szemraj, J.; Pietras, T. The Coexistence of Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. J. Clin. Med. 2022, 11, 1375. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Kobayashi, T.; Honda, Y.; Ogawa, Y.; Kessoku, T.; Imajo, K.; Nogami, A.; Taguri, M.; Kirikoshi, H.; Saito, S.; et al. Combination of Tofogliflozin and Pioglitazone for NAFLD: Extension to the ToPiND Randomized Controlled Trial. Hepatol. Commun. 2022, 6, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Muzica, C.M.; Sfarti, C.; Trifan, A.; Zenovia, S.; Cuciureanu, T.; Nastasa, R.; Huiban, L.; Cojocariu, C.; Singeap, A.-M.; Girleanu, I.; et al. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: A Bidirectional Relationship. Can. J. Gastroenterol. Hepatol. 2020, 2020, 6638306. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef] [PubMed]
- Kosmalski, M.; Śliwińska, A.; Drzewoski, J. Non-Alcoholic Fatty Liver Disease or Type 2 Diabetes Mellitus—The Chicken or the Egg Dilemma. Biomedicines 2023, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; De Michieli, F.; Paschetta, E.; Pinach, S.; Saba, F.; Bongiovanni, D.; Framarin, L.; Berrutti, M.; Leone, N.; et al. MERTK Rs4374383 Variant Predicts Incident Nonalcoholic Fatty Liver Disease and Diabetes: Role of Mononuclear Cell Activation and Adipokine Response to Dietary Fat. Hum. Mol. Genet. 2017, 26, 1747–1758. [Google Scholar] [CrossRef] [Green Version]
- Saponaro, C.; Gaggini, M.; Gastaldelli, A. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes: Common Pathophysiologic Mechanisms. Curr. Diabetes Rep. 2015, 15, 607. [Google Scholar] [CrossRef]
- Solis-Herrera, C.; Triplitt, C.; Cersosimo, E.; DeFronzo, R.A. Pathogenesis of Type 2 Diabetes Mellitus. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Akshintala, D.; Chugh, R.; Amer, F.; Cusi, K. Nonalcoholic Fatty Liver Disease: The Overlooked Complication of Type 2 Diabetes. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Freeman, A.M.; Pennings, N. Insulin Resistance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and Diabetes Mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Williams, K.H.; Shackel, N.A.; Gorrell, M.D.; McLennan, S.V.; Twigg, S.M. Diabetes and Nonalcoholic Fatty Liver Disease: A Pathogenic Duo. Endocr. Rev. 2013, 34, 84–129. [Google Scholar] [CrossRef]
- Valenti, L.; Bugianesi, E.; Pajvani, U.; Targher, G. Nonalcoholic Fatty Liver Disease: Cause or Consequence of Type 2 Diabetes? Liver Int. 2016, 36, 1563–1579. [Google Scholar] [CrossRef]
- Yoneda, M.; Honda, Y.; Ogawa, Y.; Kessoku, T.; Kobayashi, T.; Imajo, K.; Ozaki, A.; Nogami, A.; Taguri, M.; Yamanaka, T.; et al. Comparing the Effects of Tofogliflozin and Pioglitazone in Non-Alcoholic Fatty Liver Disease Patients with Type 2 Diabetes Mellitus (ToPiND Study): A Randomized Prospective Open-Label Controlled Trial. BMJ Open Diabetes Res. Care 2021, 9, e001990. [Google Scholar] [CrossRef]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pract. 2020, 35, 72–84. [Google Scholar] [CrossRef]
- Nassir, F. NAFLD: Mechanisms, Treatments, and Biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Genetic Predisposition in Nonalcoholic Fatty Liver Disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Pirola, C.J.; Sookoian, S.; Wilson, L.A.; Belt, P.; Liang, T.; Liu, W.; Chalasani, N. Impact of the Association Between PNPLA3 Genetic Variation and Dietary Intake on the Risk of Significant Fibrosis in Patients with NAFLD. Am. J. Gastroenterol. 2021, 116, 994–1006. [Google Scholar] [CrossRef]
- Tinajero, M.G.; Malik, V.S. An Update on the Epidemiology of Type 2 Diabetes. Endocrinol. Metab. Clin. N. Am. 2021, 50, 337–355. [Google Scholar] [CrossRef]
- Ahmed, S.A.H.; Ansari, S.A.; Mensah-Brown, E.P.K.; Emerald, B.S. The Role of DNA Methylation in the Pathogenesis of Type 2 Diabetes Mellitus. Clin. Epigenet. 2020, 12, 104. [Google Scholar] [CrossRef]
- Yang, B.T.; Dayeh, T.A.; Kirkpatrick, C.L.; Taneera, J.; Kumar, R.; Groop, L.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Insulin Promoter DNA Methylation Correlates Negatively with Insulin Gene Expression and Positively with HbA1c Levels in Human Pancreatic Islets. Diabetologia 2011, 54, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellulu, M.S.; Samouda, H. Clinical and Biological Risk Factors Associated with Inflammation in Patients with Type 2 Diabetes Mellitus. BMC Endocr. Disord. 2022, 22, 16. [Google Scholar] [CrossRef]
- Barbu, E.; Popescu, M.-R.; Popescu, A.-C.; Balanescu, S.-M. Inflammation as A Precursor of Atherothrombosis, Diabetes and Early Vascular Aging. Int. J. Mol. Sci. 2022, 23, 963. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.L.; Laight, D.; Aspinall, R.J.; Higginson, A.; Cummings, M.H. A Randomised Placebo Controlled Trial of VSL#3® Probiotic on Biomarkers of Cardiovascular Risk and Liver Injury in Non-Alcoholic Fatty Liver Disease. BMC Gastroenterol. 2021, 21, 144. [Google Scholar] [CrossRef]
- Haidari, F.; Hojhabrimanesh, A.; Helli, B.; Seyedian, S.-S.; Ahmadi-Angali, K. An Energy-Restricted High-Protein Diet Supplemented with β-Cryptoxanthin Alleviated Oxidative Stress and Inflammation in Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial. Nutr. Res. 2020, 73, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bansal, M.B. Role of Kupffer Cells in Driving Hepatic Inflammation and Fibrosis in HIV Infection. Front. Immunol. 2020, 11, 1086. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer Cells Promote M1 Kupffer Cell Apoptosis: A Protective Mechanism against Alcoholic and Nonalcoholic Fatty Liver Disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Wang, F.; Stappenbeck, F.; Tang, L.-Y.; Zhang, Y.E.; Hui, S.T.; Lusis, A.J.; Parhami, F. Oxy210, a Semi-Synthetic Oxysterol, Exerts Anti-Inflammatory Effects in Macrophages via Inhibition of Toll-like Receptor (TLR) 4 and TLR2 Signaling and Modulation of Macrophage Polarization. Int. J. Mol. Sci. 2022, 23, 5478. [Google Scholar] [CrossRef]
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 5381. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, Y.; Han, F.; Chang, Y.; Li, X.; Han, Z.; Xue, M.; Cheng, Y.; Sun, B.; Chen, L. Saxagliptin Regulates M1/M2 Macrophage Polarization via CaMKKβ/AMPK Pathway to Attenuate NAFLD. Biochem. Biophys. Res. Commun. 2018, 503, 1618–1624. [Google Scholar] [CrossRef]
- Feng, X.; Qin, H.; Shi, Q.; Zhang, Y.; Zhou, F.; Wu, H.; Ding, S.; Niu, Z.; Lu, Y.; Shen, P. Chrysin Attenuates Inflammation by Regulating M1/M2 Status via Activating PPARγ. Biochem. Pharmacol. 2014, 89, 503–514. [Google Scholar] [CrossRef]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver Fibrosis and Hepatic Stellate Cells: Etiology, Pathological Hallmarks and Therapeutic Targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Peters, K.M.; Wilson, R.B.; Borradaile, N.M. Non-Parenchymal Hepatic Cell Lipotoxicity and the Coordinated Progression of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 417–422. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J.A.; Li, H.; Jo, S.-E.; Lee, H.; Kim, T.H.; Kim, M.; Kim, S.-J.; Suh, S.H. Anti-Inflammatory and Anti-Fibrotic Effects of Modafinil in Nonalcoholic Liver Disease. Biomed. Pharmacother. 2021, 144, 112372. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines With Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-K. Effect of Danshao Huaxian Capsule on Gremlin and Bone Morphogenetic Protein-7 Expression in Hepatic Fibrosis in Rats. World J. Gastroenterol. 2014, 20, 14875. [Google Scholar] [CrossRef]
- Weng, H.L. Animal Experiment and Clinical Study of Effect of Gamma-Interferon on Hepatic Fibrosis. World J. Gastroenterol. 2001, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, K.H.; Kim, H.-K.; Kim, M.-J.; Back, S.H.; Konishi, M.; Itoh, N.; Lee, M.-S. Fibroblast Growth Factor 21 Participates in Adaptation to Endoplasmic Reticulum Stress and Attenuates Obesity-Induced Hepatic Metabolic Stress. Diabetologia 2015, 58, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, C.P.M.S.; Da Costa Gayotto, L.C.; Tatai, C.; Della Bina, B.I.; Janiszewski, M.; Lima, E.S.; Abdalla, D.S.P.; Lopasso, F.P.; Laurindo, F.R.M.; Laudanna, A.A. Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease, in Rats Fed with a Choline-Deficient Diet. J. Cell. Mol. Med. 2002, 6, 399–406. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Araya, J.; Poniachik, J. Oxidative Stress and Depletion of Hepatic Long-Chain Polyunsaturated Fatty Acids May Contribute to Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2004, 37, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Barrow, F.; Khan, S.; Wang, H.; Revelo, X.S. The Emerging Role of B Cells in the Pathogenesis of NAFLD. Hepatology 2021, 74, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte Responses to Oxidative Stress-Derived Antigens Contribute to the Evolution of Nonalcoholic Fatty Liver Disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Albano, E. Immune Response towards Lipid Peroxidation Products as a Predictor of Progression of Non-Alcoholic Fatty Liver Disease to Advanced Fibrosis. Gut 2005, 54, 987–993. [Google Scholar] [CrossRef]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD Progression: A Lipidomics Approach to an Epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Cao, G.; Min, X.; Wang, T.; Sun, S.; Du, X.; Zhang, W. Cathepsin B Inhibition Ameliorates the Non-Alcoholic Steatohepatitis through Suppressing Caspase-1 Activation. J. Physiol. Biochem. 2018, 74, 503–510. [Google Scholar] [CrossRef]
- Xu, G.; Fu, S.; Zhan, X.; Wang, Z.; Zhang, P.; Shi, W.; Qin, N.; Chen, Y.; Wang, C.; Niu, M.; et al. Echinatin Effectively Protects against NLRP3 Inflammasome–Driven Diseases by Targeting HSP90. JCI Insight 2021, 6, e134601. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chang, P.-F.; Chang, M.-H.; Ni, Y.-H. Genetic Variants in GCKR and PNPLA3 Confer Susceptibility to Nonalcoholic Fatty Liver Disease in Obese Individuals. Am. J. Clin. Nutr. 2014, 99, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-C.; Chang, P.-F.; Chang, M.-H.; Ni, Y.-H. A Common Variant in the Peroxisome Proliferator–Activated Receptor-γ Coactivator-1α Gene Is Associated with Nonalcoholic Fatty Liver Disease in Obese Children. Am. J. Clin. Nutr. 2013, 97, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma Transforming Growth Factor-Β1 Level and Efficacy of α-Tocopherol in Patients with Non-Alcoholic Steatohepatitis: A Pilot Study: TOCOPHEROL FOR NASH. Aliment. Pharmacol. Ther. 2001, 15, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; Van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic Recruitment of Macrophages Promotes Nonalcoholic Steatohepatitis through CCR2. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiman, Y.; Friedman, S.L. The Role of Chemokines in Acute Liver Injury. Front. Physiol. 2012, 3, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocsan, I.C.; Milaciu, M.V.; Pop, R.M.; Vesa, S.C.; Ciumarnean, L.; Matei, D.M.; Buzoianu, A.D. Cytokines Genotype-Phenotype Correlation in Nonalcoholic Steatohepatitis. Oxidative Med. Cell. Longev. 2017, 2017, 4297206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amerikanou, C.; Papada, E.; Gioxari, A.; Smyrnioudis, I.; Kleftaki, S.-A.; Valsamidou, E.; Bruns, V.; Banerjee, R.; Trivella, M.G.; Milic, N.; et al. Mastiha Has Efficacy in Immune-Mediated Inflammatory Diseases through a MicroRNA-155 Th17 Dependent Action. Pharmacol. Res. 2021, 171, 105753. [Google Scholar] [CrossRef]
- Cangeri Di Naso, F.; Rosa Porto, R.; Sarubbi Fillmann, H.; Maggioni, L.; Vontobel Padoin, A.; Jacques Ramos, R.; Corá Mottin, C.; Bittencourt, A.; Anair Possa Marroni, N.; Ivo Homem de Bittencourt, P. Obesity Depresses the Anti-Inflammatory HSP70 Pathway, Contributing to NAFLD Progression: Obesity Depresses HSP70 in NAFLD. Obesity 2015, 23, 120–129. [Google Scholar] [CrossRef]
- El-Kader, S.M.A.; Al-Shreef, F.M.; Al-Jiffri, O.H. Biochemical Parameters Response to Weight Loss in Patients with Non-Alcoholic Steatohepatitis. Afr. Health Sci. 2016, 16, 242. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Yoshio, S.; Doi, H.; Mori, T.; Matsuda, M.; Kawai, H.; Shimagaki, T.; Yoshikawa, S.; Aoki, Y.; Osawa, Y.; et al. Increased Frequency of Dysfunctional Siglec-7−CD57+PD-1+ Natural Killer Cells in Patients with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2021, 12, 603133. [Google Scholar] [CrossRef]
- Cifarelli, V.; Beeman, S.C.; Smith, G.I.; Yoshino, J.; Morozov, D.; Beals, J.W.; Kayser, B.D.; Watrous, J.D.; Jain, M.; Patterson, B.W.; et al. Decreased Adipose Tissue Oxygenation Associates with Insulin Resistance in Individuals with Obesity. J. Clin. Investig. 2020, 130, 6688–6699. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, R.; Yang, X.; Dai, J.; Huang, M.; Ji, X.; Li, Y.; Okekunle, A.P.; Gao, G.; Onwuka, J.U.; et al. Yogurt Improves Insulin Resistance and Liver Fat in Obese Women with Nonalcoholic Fatty Liver Disease and Metabolic Syndrome: A Randomized Controlled Trial. Am. J. Clin. Nutr. 2019, 109, 1611–1619. [Google Scholar] [CrossRef]
- Khalyfa, A.; Gozal, D.; Masa, J.F.; Marin, J.M.; Qiao, Z.; Corral, J.; González, M.; Marti, S.; Kheirandish-Gozal, L.; Egea, C.; et al. Sleep-Disordered Breathing, Circulating Exosomes, and Insulin Sensitivity in Adipocytes. Int. J. Obes. 2018, 42, 1127–1139. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Reversal of Insulin Resistance in Overweight and Obese Subjects by Trans-Resveratrol and Hesperetin Combination—Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, T.M.; Seelig, E.; Timper, K.; Cordes, M.; Coslovsky, M.; Hanssen, H.; Schmidt-Trucksäss, A.; Donath, M.Y. Muscle-Derived IL-6 Is Not Regulated by IL-1 during Exercise. A Double Blind, Placebo-Controlled, Randomized Crossover Study. PLoS ONE 2015, 10, e0139662. [Google Scholar] [CrossRef] [Green Version]
- Benrick, A.; Wallenius, V.; Asterholm, I.W. Interleukin-6 Mediates Exercise-Induced Increase in Insulin Sensitivity in Mice. Exp. Physiol. 2012, 97, 1224–1235. [Google Scholar] [CrossRef]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 Promotes Alternative Activation of Macrophages to Limit Endotoxemia and Obesity-Associated Resistance to Insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H.; Liaqat, A.; Kamal, S.; Qadir, M.I.; Rasul, A. Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.-F.; Lu, D.-H.; Liu, H.-L.; Liu, Y.-J.; Han, X.-Q.; Yang, Y.; Lin, Y.; Zeng, Q.-X.; Huang, Z.-J.; Xie, F.; et al. Effectiveness and Safety of Human Umbilical Cord-Mesenchymal Stem Cells for Treating Type 2 Diabetes Mellitus. World J. Diabetes 2022, 13, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of Type 2 Diabetes: Normalisation of Beta Cell Function in Association with Decreased Pancreas and Liver Triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [Green Version]
- Noe, A.; Howard, C.; Thuren, T.; Taylor, A.; Skerjanec, A. Pharmacokinetic and Pharmacodynamic Characteristics of Single-Dose Canakinumab in Patients with Type 2 Diabetes Mellitus. Clin. Ther. 2014, 36, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- van Poppel, P.C.M.; van Asseldonk, E.J.P.; Holst, J.J.; Vilsbøll, T.; Netea, M.G.; Tack, C.J. The Interleukin-1 Receptor Antagonist Anakinra Improves First-Phase Insulin Secretion and Insulinogenic Index in Subjects with Impaired Glucose Tolerance. Diabetes Obes. Metab. 2014, 16, 1269–1273. [Google Scholar] [CrossRef]
- Larsen, C.M.; Vølund, A.; Ehses, J.A.; Mandrup-Poulsen, T. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Rissanen, A.; Howard, C.P.; Botha, J.; Thuren, T.; for the Global Investigators. Effect of Anti-IL-1β Antibody (Canakinumab) on Insulin Secretion Rates in Impaired Glucose Tolerance or Type 2 Diabetes: Results of a Randomized, Placebo-Controlled Trial. Diabetes Obes. Metab. 2012, 14, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-T.; Xiong, D.-Y.; Xiao, J.-N.; Deng, L.; Liu, W.; Tang, S.-Y. Kindlin-2 Protects Pancreatic β Cells through Inhibiting NLRP3 Inflammasome Activation in Diabetic Mice. Biochem. Biophys. Res. Commun. 2022, 614, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yan, Z.; Cheng, Y.; Zhong, M.; Liu, S.; Zhang, G.; Hu, S. Deactivation of the NLRP3 Inflammasome in Infiltrating Macrophages by Duodenal-Jejunal Bypass Surgery Mediates Improvement of Beta Cell Function in Type 2 Diabetes. Metabolism 2018, 81, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wali, J.A.; Gurzov, E.N.; Fynch, S.; Elkerbout, L.; Kay, T.W.; Masters, S.L.; Thomas, H.E. Activation of the NLRP3 Inflammasome Complex Is Not Required for Stress-Induced Death of Pancreatic Islets. PLoS ONE 2014, 9, e113128. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wang, Y.; Guo, M.; Long, Y.; Chen, J.; Fan, F.; Tang, S.; Xu, Y. PCB118 Induces Inflammation of Islet Beta Cells via Activating ROS-NLRP3 Inflammasome Signaling. BioMed Res. Int. 2021, 2021, 5522578. [Google Scholar] [CrossRef]
- Abderrazak, A.; El Hadri, K.; Bosc, E.; Blondeau, B.; Slimane, M.-N.; Buchele, B.; Simmet, T.; Couchie, D.; Rouis, M. Inhibition of the Inflammasome NLRP3 by Arglabin Attenuates Inflammation, Protects Pancreatic-Cells from Apoptosis, and Prevents Type 2 Diabetes Mellitus Development in ApoE2Ki Mice on a Chronic High-Fat Diet. J. Pharmacol. Exp. Ther. 2016, 357, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lu, F.; Li, L.; Li, J.; Luo, J.; Zhang, S.; Liu, X.; Chen, G. Wu-Mei-Wan Protects Pancreatic β Cells by Inhibiting NLRP3 Inflammasome Activation in Diabetic Mice. BMC Complement. Altern. Med. 2019, 19, 35. [Google Scholar] [CrossRef] [Green Version]
- Ellingsgaard, H.; Seelig, E.; Timper, K.; Coslovsky, M.; Soederlund, L.; Lyngbaek, M.P.; Wewer Albrechtsen, N.J.; Schmidt-Trucksäss, A.; Hanssen, H.; Frey, W.O.; et al. GLP-1 Secretion Is Regulated by IL-6 Signalling: A Randomised, Placebo-Controlled Study. Diabetologia 2020, 63, 362–373. [Google Scholar] [CrossRef]
- Lang Lehrskov, L.; Lyngbaek, M.P.; Soederlund, L.; Legaard, G.E.; Ehses, J.A.; Heywood, S.E.; Wewer Albrechtsen, N.J.; Holst, J.J.; Karstoft, K.; Pedersen, B.K.; et al. Interleukin-6 Delays Gastric Emptying in Humans with Direct Effects on Glycemic Control. Cell Metab. 2018, 27, 1201–1211.e3. [Google Scholar] [CrossRef] [Green Version]
- Dauriz, M.; Trombetta, M.; Boselli, L.; Santi, L.; Brangani, C.; Pichiri, I.; Bonora, E.; Bonadonna, R.C. Interleukin-6 as a Potential Positive Modulator of Human Beta-Cell Function: An Exploratory Analysis—The Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 6. Acta Diabetol. 2016, 53, 393–402. [Google Scholar] [CrossRef]
- Weber, K.S.; Nowotny, B.; Strassburger, K.; Pacini, G.; Müssig, K.; Szendroedi, J.; Herder, C.; Roden, M.; for the GDS Group. The Role of Markers of Low-Grade Inflammation for the Early Time Course of Glycemic Control, Glucose Disappearance Rate, and β-Cell Function in Recently Diagnosed Type 1 and Type 2 Diabetes. Diabetes Care 2015, 38, 1758–1767. [Google Scholar] [CrossRef] [Green Version]
- Tuomainen, M.; Lindström, J.; Lehtonen, M.; Auriola, S.; Pihlajamäki, J.; Peltonen, M.; Tuomilehto, J.; Uusitupa, M.; de Mello, V.D.; Hanhineva, K. Associations of Serum Indolepropionic Acid, a Gut Microbiota Metabolite, with Type 2 Diabetes and Low-Grade Inflammation in High-Risk Individuals. Nutr. Diabetes 2018, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsogiannos, P.; Kamble, P.G.; Pereira, M.J.; Sundbom, M.; Carlsson, P.; Eriksson, J.W.; Espes, D. Changes in Circulating Cytokines and Adipokines After RYGB in Patients with and without Type 2 Diabetes. Obesity 2021, 29, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Porter Starr, K.N.; Orenduff, M.; McDonald, S.R.; Mulder, H.; Sloane, R.; Pieper, C.F.; Bales, C.W. Influence of Weight Reduction and Enhanced Protein Intake on Biomarkers of Inflammation in Older Adults with Obesity. J. Nutr. Gerontol. Geriatr. 2019, 38, 33–49. [Google Scholar] [CrossRef]
- Dominguez, H.; Storgaard, H.; Rask-Madsen, C.; Steffen Hermann, T.; Ihlemann, N.; Baunbjerg Nielsen, D.; Spohr, C.; Kober, L.; Vaag, A.; Torp-Pedersen, C. Metabolic and Vascular Effects of Tumor Necrosis Factor-α Blockade with Etanercept in Obese Patients with Type 2 Diabetes. J. Vasc. Res. 2005, 42, 517–525. [Google Scholar] [CrossRef]
- Ibfelt, T.; Fischer, C.P.; Plomgaard, P.; van Hall, G.; Pedersen, B.K. The Acute Effects of Low-Dose TNF-α on Glucose Metabolism and β-Cell Function in Humans. Mediat. Inflamm. 2014, 2014, 295478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.J.; James, L.; Hussey, B.; Wadley, A.J.; Lindley, M.R.; Mastana, S.S. Impact of Aerobic Exercise and Fatty Acid Supplementation on Global and Gene-Specific DNA Methylation. Epigenetics 2019, 14, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Lucia, A.; Gómez-Gallego, F.; Barroso, I.; Rabadán, M.; Bandrés, F.; San Juan, A.F.; Chicharro, J.L.; Ekelund, U.; Brage, S.; Earnest, C.P.; et al. PPARGC1A Genotype (Gly482Ser) Predicts Exceptional Endurance Capacity in European Men. J. Appl. Physiol. 2005, 99, 344–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensink, M.; Hesselink, M.; Russell, A.; Schaart, G.; Sels, J.-P.; Schrauwen, P. Improved Skeletal Muscle Oxidative Enzyme Activity and Restoration of PGC-1a and PPARb/d Gene Expression upon Rosiglitazone Treatment in Obese Patients with Type 2 Diabetes Mellitus. Int. J. Obes. 2007, 31, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Alvarez, M.I.; Thabit, H.; Burns, N.; Shah, S.; Brema, I.; Hatunic, M.; Finucane, F.; Liesa, M.; Chiellini, C.; Naon, D.; et al. Subjects with Early-Onset Type 2 Diabetes Show Defective Activation of the Skeletal Muscle PGC-1α/Mitofusin-2 Regulatory Pathway in Response to Physical Activity. Diabetes Care 2010, 33, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Mo, W.; Feng, J.; Li, J.; Yu, Q.; Li, S.; Zhang, J.; Chen, K.; Ji, J.; Dai, W.; et al. Astaxanthin Attenuates Hepatic Damage and Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease by Up-regulating the FGF21/PGC-1α Pathway. Br. J. Pharm. 2020, 177, 3760–3777. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Chen, M. Progress in Nonalcoholic Fatty Liver Disease: SIRT Family Regulates Mitochondrial Biogenesis. Biomolecules 2022, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwarkanath, A.; Dwivedi, U.N.; Kakkar, P. Berbamine Induced Activation of the SIRT1/LKB1/AMPK Signaling Axis Attenuates the Development of Hepatic Steatosis in High-Fat Diet-Induced NAFLD Rats. Food Funct. 2021, 12, 892–909. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Wu, C.-L.; Su, W.-W.; Shih, K.-L.; Tarng, D.-C.; Chou, C.-T.; Chen, T.-Y.; Kor, C.-T.; Wu, H.-M. Interferon Gamma-Induced Protein 10 Is Associated with Insulin Resistance and Incident Diabetes in Patients with Nonalcoholic Fatty Liver Disease. Sci. Rep. 2015, 5, 10096. [Google Scholar] [CrossRef] [Green Version]
- Barchetta, I.; Cimini, F.A.; De Gioannis, R.; Ciccarelli, G.; Bertoccini, L.; Lenzi, A.; Baroni, M.G.; Cavallo, M.G. Procollagen-III Peptide Identifies Adipose Tissue-Associated Inflammation in Type 2 Diabetes with or without Nonalcoholic Liver Disease. Diabetes Metab. Res. Rev. 2018, 34, e2998. [Google Scholar] [CrossRef]
- Osorio-Conles, Ó.; Vega-Beyhart, A.; Ibarzabal, A.; Balibrea, J.M.; Graupera, I.; Rimola, J.; Vidal, J.; de Hollanda, A. A Distinctive NAFLD Signature in Adipose Tissue from Women with Severe Obesity. Int. J. Mol. Sci. 2021, 22, 10541. [Google Scholar] [CrossRef]
- Lê, K.-A.; Mahurkar, S.; Alderete, T.L.; Hasson, R.E.; Adam, T.C.; Kim, J.S.; Beale, E.; Xie, C.; Greenberg, A.S.; Allayee, H.; et al. Subcutaneous Adipose Tissue Macrophage Infiltration Is Associated with Hepatic and Visceral Fat Deposition, Hyperinsulinemia, and Stimulation of NF-ΚB Stress Pathway. Diabetes 2011, 60, 2802–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magkos, F.; Fraterrigo, G.; Yoshino, J.; Luecking, C.; Kirbach, K.; Kelly, S.C.; de las Fuentes, L.; He, S.; Okunade, A.L.; Patterson, B.W.; et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 2016, 23, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Berings, M.; Wehlou, C.; Verrijken, A.; Deschepper, E.; Mertens, I.; Kaufman, J.-M.; Van Gaal, L.F.; Ouwens, D.M.; Ruige, J.B. Glucose Intolerance and the Amount of Visceral Adipose Tissue Contribute to an Increase in Circulating Triglyceride Concentrations in Caucasian Obese Females. PLoS ONE 2012, 7, e45145. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like Peptide 1 Decreases Lipotoxicity in Non-Alcoholic Steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenette, C.; Kayali, Z.; Mena, E.; Mantry, P.S.; Lucas, K.J.; Neff, G.; Rodriguez, M.; Thuluvath, P.J.; Weinberg, E.; Bhandari, B.R.; et al. Emricasan to Prevent New Decompensation in Patients with NASH-Related Decompensated Cirrhosis. J. Hepatol. 2021, 74, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Kashyap, S.; Gastaldelli, A.; Bajaj, M.; Cersosimo, E. Effects on Insulin Secretion and Insulin Action of a 48-h Reduction of Plasma Free Fatty Acids with Acipimox in Nondiabetic Subjects Genetically Predisposed to Type 2 Diabetes. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E1775–E1781. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin Resistance Drives Hepatic de Novo Lipogenesis in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Ramírez, Á.; Hernández, M.; Suárez-Sánchez, R.; Ortega, C.; Peralta, J.; Gómez, J.; Valladares, A.; Cruz, M.; Vázquez-Moreno, M.A.; Suárez-Sánchez, F. Type 2 Diabetes–Associated Polymorphisms Correlate with SIRT1 and TGF-β1 Gene Expression. Ann. Hum. Genet. 2020, 84, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Honda, Y.; Kurita, Y.; Iwasaki, A.; Sato, T.; Kessoku, T.; Uchiyama, S.; Ogawa, Y.; Ohkubo, H.; Higurashi, T.; et al. Lubiprostone Improves Intestinal Permeability in Humans, a Novel Therapy for the Leaky Gut: A Prospective Randomized Pilot Study in Healthy Volunteers. PLoS ONE 2017, 12, e0175626. [Google Scholar] [CrossRef] [Green Version]
- Farrokhian, A.; Raygan, F.; Soltani, A.; Tajabadi-Ebrahimi, M.; Sharifi Esfahani, M.; Karami, A.A.; Asemi, Z. The Effects of Synbiotic Supplementation on Carotid Intima-Media Thickness, Biomarkers of Inflammation, and Oxidative Stress in People with Overweight, Diabetes, and Coronary Heart Disease: A Randomized, Double-Blind, Placebo-Controlled Trial. Probiotics Antimicro. Prot. 2019, 11, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Nor, M.H.; Ayob, N.; Mokhtar, N.M.; Raja Ali, R.A.; Tan, G.C.; Wong, Z.; Shafiee, N.H.; Wong, Y.P.; Mustangin, M.; Nawawi, K.N.M. The Effect of Probiotics (MCP® BCMC® Strains) on Hepatic Steatosis, Small Intestinal Mucosal Immune Function, and Intestinal Barrier in Patients with Non-Alcoholic Fatty Liver Disease. Nutrients 2021, 13, 3192. [Google Scholar] [CrossRef]
- Shen, L.; Ao, L.; Xu, H.; Shi, J.; You, D.; Yu, X.; Xu, W.; Sun, J.; Wang, F. Poor Short-Term Glycemic Control in Patients with Type 2 Diabetes Impairs the Intestinal Mucosal Barrier: A Prospective, Single-Center, Observational Study. BMC Endocr. Disord. 2019, 19, 29. [Google Scholar] [CrossRef] [Green Version]
- Hietbrink, F.; Besselink, M.G.H.; Renooij, W.; de Smet, M.B.M.; Draisma, A.; van der Hoeven, H.; Pickkers, P. Systemic inflammation increases intestinal permeability during experimental human endotoxemia. Shock 2009, 32, 374–378. [Google Scholar] [CrossRef]
- Shen, H.; Shahzad, G.; Jawairia, M.; Bostick, R.M.; Mustacchia, P. Association between Aspirin Use and the Prevalence of Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study from the Third National Health and Nutrition Examination Survey. Aliment Pharm. Ther. 2014, 40, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Khajehdehi, P.; Roozbeh, J.; Mostafavi, H. A Comparative Randomized and Placebo-Controlled Short-Term Trial of Aspirin and Dipyridamole for Overt Type-2 Diabetic Nephropathy. Scand. J. Urol. Nephrol. 2002, 36, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Parolini, M. Toxicity of the Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) Acetylsalicylic Acid, Paracetamol, Diclofenac, Ibuprofen and Naproxen towards Freshwater Invertebrates: A Review. Sci. Total Environ. 2020, 740, 140043. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, H.; Liu, Z.; Zhang, K.K.; Jendrusch, C.; Drake, M.; Hao, Y.; Xie, L. Sex-Associated Preventive Effects of Low-Dose Aspirin on Obesity and Non-Alcoholic Fatty Liver Disease in Mouse Offspring with over-Nutrition in Utero. Lab. Investig. 2019, 99, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.G.; Feldbrügge, L.; Tapper, E.B.; Popov, Y.; Ghaziani, T.; Afdhal, N.; Robson, S.C.; Mukamal, K.J. Aspirin Use Is Associated with Lower Indices of Liver Fibrosis among Adults in the United States. Aliment Pharm. Ther. 2016, 43, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, C.; Ogawa, H.; Saito, Y.; Okada, S.; Soejima, H.; Sakuma, M.; Masuda, I.; Nakayama, M.; Doi, N.; Jinnouchi, H.; et al. Sex Difference in Effects of Low-Dose Aspirin on Prevention of Dementia in Patients With Type 2 Diabetes: A Long-Term Follow-up Study of a Randomized Clinical Trial. Diabetes Care 2020, 43, 314–320. [Google Scholar] [CrossRef]
- Lin, M.-H.; Wu, W.-T.; Chen, Y.-C.; Lu, C.-H.; Su, S.-C.; Kuo, F.-C.; Chou, Y.-C.; Sun, C.-A. Association between Non-Steroidal Anti-Inflammatory Drugs Use and the Risk of Type 2 Diabetes Mellitus: A Nationwide Retrospective Cohort Study. J. Clin. Med. 2022, 11, 3186. [Google Scholar] [CrossRef]
- Nakamura, Y.; Tsuji, M.; Hasegawa, H.; Kimura, K.; Fujita, K.; Inoue, M.; Shimizu, T.; Gotoh, H.; Goto, Y.; Inagaki, M.; et al. Anti-Inflammatory Effects of Linagliptin in Hemodialysis Patients with Diabetes: Linagliptin Therapy in Dialysis Patients. Hemodial. Int. 2014, 18, 433–442. [Google Scholar] [CrossRef]
- Seo, H.-Y.; Lee, S.-H.; Han, E.; Hwang, J.S.; Han, S.; Kim, M.K.; Jang, B.K. Evogliptin Directly Inhibits Inflammatory and Fibrotic Signaling in Isolated Liver Cells. Int. J. Mol. Sci. 2022, 23, 11636. [Google Scholar] [CrossRef]
- Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; Tuin, S.J.L.; Zhang, H.; Bieghs, V.; Jawad, A.H.M.; Shiri-Sverdlov, R.; Bot, I.; Jager, S.C.A.; et al. Exendin-4 Decreases Liver Inflammation and Atherosclerosis Development Simultaneously by Reducing Macrophage Infiltration. Br. J. Pharm. 2014, 171, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Krysiak, R.; Gdula-Dymek, A.; Okopien, B. Effect of Simvastatin and Fenofibrate on Cytokine Release and Systemic Inflammation in Type 2 Diabetes Mellitus with Mixed Dyslipidemia. Am. J. Cardiol. 2011, 107, 1010–1018.e1. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Nikolajczyk, B.S. The Intersection of Metformin and Inflammation. Am. J. Physiol. Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.M.; Donath, M.Y.; LeRoith, D.; Leibowitz, G. Anti-Inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39 (Suppl. 2), S244–S252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A.J. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-Activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, S.; Houseright, R.A.; Graves, A.L.; Golenberg, N.; Korte, B.G.; Miskolci, V.; Huttenlocher, A. Metformin Modulates Innate Immune-Mediated Inflammation and Early Progression of NAFLD-Associated Hepatocellular Carcinoma in Zebrafish. J. Hepatol. 2019, 70, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Mitrovic, B.; Gluvic, Z.; Macut, D.; Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Stajic, D.; Isenovic, E.R. Effects of Metformin-Single Therapy on the Level of Inflammatory Markers in Serum of Non-Obese T2DM Patients with NAFLD. Endocr. Metab. Immune Disord.-Drug Targets 2022, 22, 117–124. [Google Scholar] [CrossRef]
- La Grotta, R.; de Candia, P.; Olivieri, F.; Matacchione, G.; Giuliani, A.; Rippo, M.R.; Tagliabue, E.; Mancino, M.; Rispoli, F.; Ferroni, S.; et al. Anti-Inflammatory Effect of SGLT-2 Inhibitors via Uric Acid and Insulin. Cell. Mol. Life Sci. 2022, 79, 273. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Z.; Wang, J.; Zhang, X.; Yu, X.; Li, Y. Empagliflozin Protects Diabetic Pancreatic Tissue from Damage by Inhibiting the Activation of the NLRP3/Caspase-1/GSDMD Pathway in Pancreatic β Cells: In Vitro and In Vivo Studies. Bioengineered 2021, 12, 9356–9366. [Google Scholar] [CrossRef]
- Takahashi, H.; Kessoku, T.; Kawanaka, M.; Nonaka, M.; Hyogo, H.; Fujii, H.; Nakajima, T.; Imajo, K.; Tanaka, K.; Kubotsu, Y.; et al. Ipragliflozin Improves the Hepatic Outcomes of Patients with Diabetes with NAFLD. Hepatol. Commun. 2022, 6, 120–132. [Google Scholar] [CrossRef]
- Leite, N.C.; Viegas, B.B.; Villela-Nogueira, C.A.; Carlos, F.O.; Cardoso, C.R.L.; Salles, G.F. Efficacy of Diacerein in Reducing Liver Steatosis and Fibrosis in Patients with Type 2 Diabetes and Non-alcoholic Fatty Liver Disease: A Randomized, Placebo-controlled Trial. Diabetes Obes. Metab. 2019, 21, 1266–1270. [Google Scholar] [CrossRef]
- Cavelti-Weder, C.; Babians-Brunner, A.; Keller, C.; Stahel, M.A.; Kurz-Levin, M.; Zayed, H.; Solinger, A.M.; Mandrup-Poulsen, T.; Dinarello, C.A.; Donath, M.Y. Effects of Gevokizumab on Glycemia and Inflammatory Markers in Type 2 Diabetes. Diabetes Care 2012, 35, 1654–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscitti, P.; Masedu, F.; Alvaro, S.; Airò, P.; Battafarano, N.; Cantarini, L.; Cantatore, F.P.; Carlino, G.; D’Abrosca, V.; Frassi, M.; et al. Anti-Interleukin-1 Treatment in Patients with Rheumatoid Arthritis and Type 2 Diabetes (TRACK): A Multicentre, Open-Label, Randomised Controlled Trial. PLoS Med. 2019, 16, e1002901. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Burmester, G.R.; Hagino, O.; Thangavelu, K.; Iglesias-Rodriguez, M.; John, G.S.; González-Gay, M.A.; Mandrup-Poulsen, T.; Fleischmann, R. Interleukin-6 Receptor Blockade or TNFα Inhibition for Reducing Glycaemia in Patients with RA and Diabetes: Post Hoc Analyses of Three Randomised, Controlled Trials. Arthritis Res. Ther. 2020, 22, 206. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Study | Group | Inflammation, NAFLD, DM2 Induction | Intervention | Duration | Outcomes |
|---|---|---|---|---|---|
| Choi et al. [78] | 110 C57BL/6 mice | Steatosis, steatohepatitis, and liver fibrosis induced by choline-deficient, L-amino-acid-defined, high-fat diet with 0.1% methionine | Modafinil in doses 10, 50, or 100 mg/kg | 20 weeks | Used diet and TAA lead to KCa2.3, KCa3.1, upregulation, and downregulation of catalase in liver tissues Modafinil can reverse KCa2.3, KCa3.1, collagen, and α-smooth muscle actin upregulation and downregulation of catalase, and leads to a decline in the inflammatory response, collagen deposition, and α-smooth muscle actin expression |
| 75 C57BL/6 male mice | Hepatitis and fibrosis induced by TAA in dose 100 mg/kg intraperitoneally 3 times per week | Modafinil in doses 10, 50, or 100 mg/kg | 16 weeks | ||
| Zhao et al. [80] | 50 male Wistar rats | Liver fibrosis induced by carbon tetrachloride CCl4 subcutaneous injections and high-lipid/low-protein diet for 8 weeks | DHC administration in doses 0.5 or 1.0 g/kg | 8 weeks | TGF-β1 and the expression of Gremlin mRNA and protein were higher than in the control group Expression of BMP-7 mRNA and protein lower than in the control group Improvement in liver fibrosis, Decrease in TGF-β1 and the mRNA and protein expression of Gremlin in DHC groups |
| Weng et al. [81] | 483 Sprague Dawley rats (205 in the CCL4 group and 278 in the DMN group) | Liver fibrosis induced by subcutaneous injection of CCl4 or intraperitoneal injection of DMN | IFN-γ administration (in different doses: 1.67 MU/kg daily, 5 MU/kg daily, and 15 MU/kg daily) | 8 weeks for the CCL4 group 4 weeks for the DMN group | IFN-γ administration decreased the HSCs activation and is effective in reducing liver fibrosis The results of IFN-γ are dose-dependent (better results are achieved with a higher dose) |
| Tang et al. [91] | 40 male C57BL/6 mice | 6-week methionine choline-deficient diet to establish NASH | 2 weeks administration of CTSB inhibitor (CA-074 methyl ester) | 6 weeks | Higher expression of CTSB and caspase-1 than in the normal diet group Possible regulation of caspase-1 levels by CTSB CTSB inhibition results in a decline in IL-1β and IL-18 levels and downregulation of NLRP3 inflammasome in KCs |
| Benrick et al. [108] | 18 IL-6−/− mice and 18 wild-type mice | High-fat diet to induce weight gain | Access to running wheels and lack of this access | 4 weeks | IL-6 contributes to the exercise-associated increase in insulin sensitivity A high-fat diet without running led to impairing insulin sensitivity; in contrast, running was a preventive factor in conditions of insulin sensitivity in wild-type but not in IL-6−/− mice |
| Huang et al. [117] | C57BL/6 mice and a mice insulinoma immortalized β-cell line MIN6 | Diabetes induced by streptozotocin in dose 40 mg/kg intraperitoneal for five days or high-fat and high-sucrose diet | AAV8 to induce expression of Kindlin-2 | 12 weeks | Insufficiency of Kindlin-2 leads to exacerbating diabetes, promotes β-cell inflammation and dysfunction induced by a high-fat diet Overexpression of Kindlin-2 improves insulin secretion and ameliorates diabetes induced by streptozotocin In vitro model of high-glucose-induced β-cell dysfunction revealed that overexpression of Kindlin-2 leads to decreased expression of proinflammatory cytokines and NLRP3 inflammasome in β cells |
| Jiang et al. [120] | Mouse islet β-TC-6 cells | PCB118 (5, 10, and 20 nmol/L) | 48 or 72 h | NLRP3 inflammasome signaling pathways in β cells are important in diabetes development | |
| Abderrazak et al. [121] | 12 ApoE2.Ki female mice | Chronic high-fat diet | Arglabin 2.5 ng/g twice a day in intraperitoneal injection | 13 weeks | Inhibition of NLRP3 caused by arglabin leads to a decrease in inflammation and apoptosis in pancreatic β cells |
| Yang et al. [122] | 24 diabetes-prone C57BLKS/J-Leprdb/Leprdb (db/db) male mice and 24 wild-type male mice | WMW in different doses (4800, 9600, and 19,200 mg/kg) | 4 weeks | Compared with the control group diabetic mice had higher protein expression levels of NLRP3 inflammasome components NLRP3 and caspase-1 (P20) than wild-type mice WMW decreases caspase-12, increases Bcl-2 expression, and decreases the upregulated production of IL-1β,IL-18,MCP-1α, and macrophage-specific surface glycoprotein F4/80 in diabetic mice | |
| Sharma et al. [138] | 36 male Wistar rats | NAFLD induced by 12 weeks high-fat diet | Berbamine (50 or 150 mg per kg) | 12 weeks + 28 days | Improvements in liver function, liver index, and liver image due to berbamine administration Inducing the SIRT1/LKB1/AMPK pathway leads to protection against hepatic lipid metabolic disorders |
| Zhou et al. [158] | Mice with maternal overnutrition | Obesity and NAFLD induced by high-fat diet + diethylnitrosamine (intraperitoneally 20–25 μg/g and 50 μg/L in drinking water at age 21 days) | 90 μg to 120 μg aspirin per day | 12 weeks | Improvement in insulin/Akt signaling, activation of AMPK signaling, inhibition of Wnt-signaling and MAPK signaling leads to improvements in glucose intolerance, weight gain, and liver fat accumulation in female mice |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frankowski, R.; Kobierecki, M.; Wittczak, A.; Różycka-Kosmalska, M.; Pietras, T.; Sipowicz, K.; Kosmalski, M. Type 2 Diabetes Mellitus, Non-Alcoholic Fatty Liver Disease, and Metabolic Repercussions: The Vicious Cycle and Its Interplay with Inflammation. Int. J. Mol. Sci. 2023, 24, 9677. https://doi.org/10.3390/ijms24119677
Frankowski R, Kobierecki M, Wittczak A, Różycka-Kosmalska M, Pietras T, Sipowicz K, Kosmalski M. Type 2 Diabetes Mellitus, Non-Alcoholic Fatty Liver Disease, and Metabolic Repercussions: The Vicious Cycle and Its Interplay with Inflammation. International Journal of Molecular Sciences. 2023; 24(11):9677. https://doi.org/10.3390/ijms24119677
Chicago/Turabian StyleFrankowski, Rafał, Mateusz Kobierecki, Andrzej Wittczak, Monika Różycka-Kosmalska, Tadeusz Pietras, Kasper Sipowicz, and Marcin Kosmalski. 2023. "Type 2 Diabetes Mellitus, Non-Alcoholic Fatty Liver Disease, and Metabolic Repercussions: The Vicious Cycle and Its Interplay with Inflammation" International Journal of Molecular Sciences 24, no. 11: 9677. https://doi.org/10.3390/ijms24119677
APA StyleFrankowski, R., Kobierecki, M., Wittczak, A., Różycka-Kosmalska, M., Pietras, T., Sipowicz, K., & Kosmalski, M. (2023). Type 2 Diabetes Mellitus, Non-Alcoholic Fatty Liver Disease, and Metabolic Repercussions: The Vicious Cycle and Its Interplay with Inflammation. International Journal of Molecular Sciences, 24(11), 9677. https://doi.org/10.3390/ijms24119677