
Improving Mitochondrial Function in Skeletal Muscle Contributes to the Amelioration of Insulin Resistance by Nicotinamide Riboside
 ,
, 
Abstract
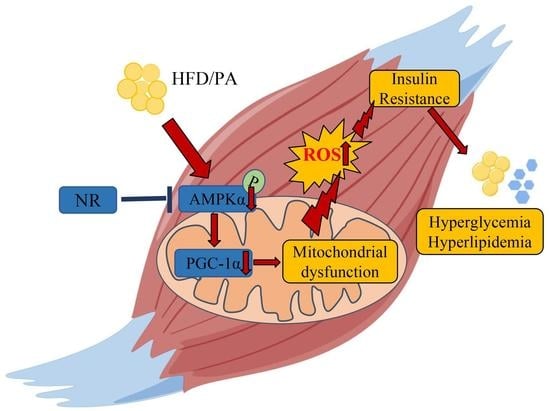
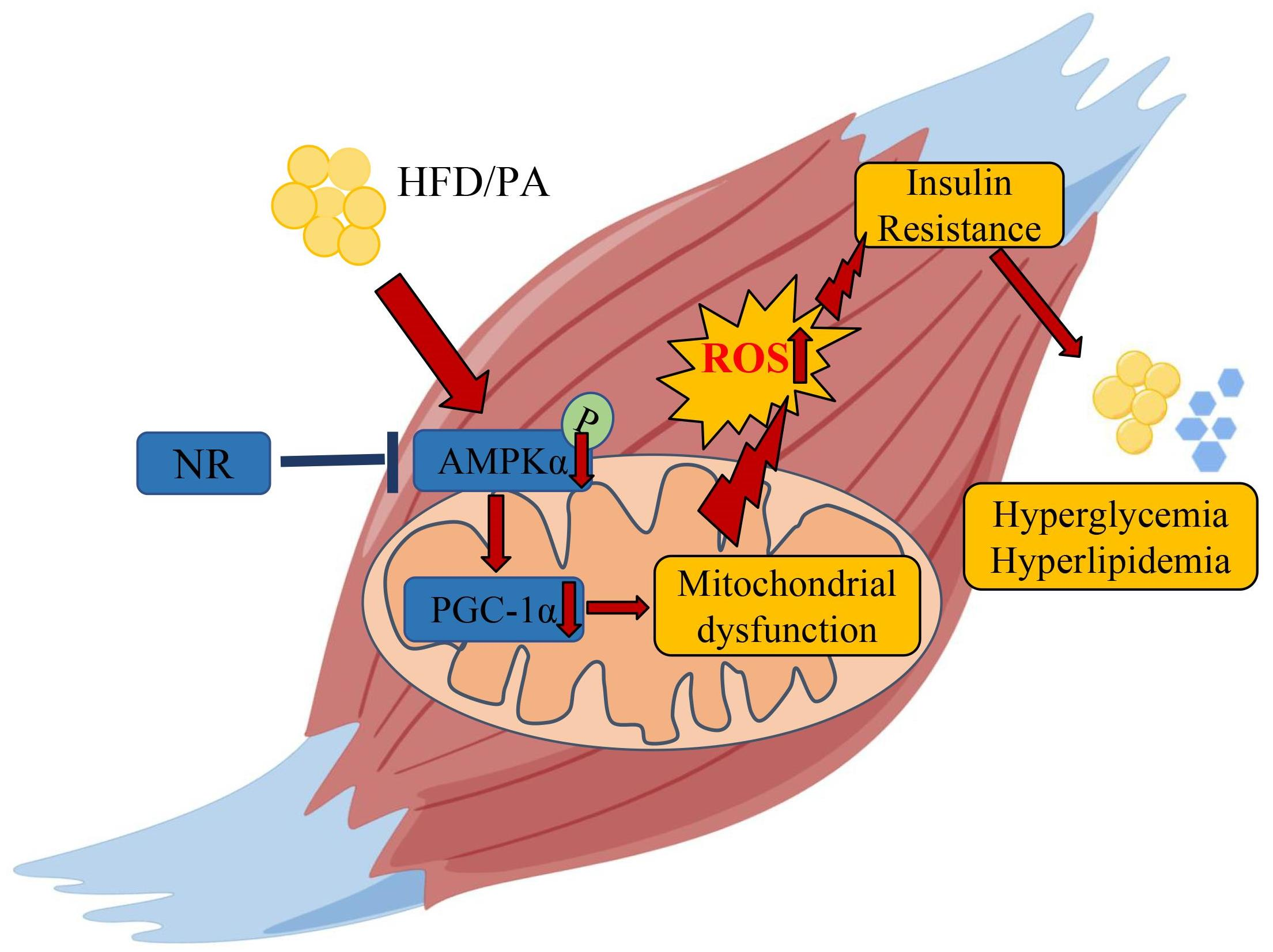
:
1. Introduction
2. Results
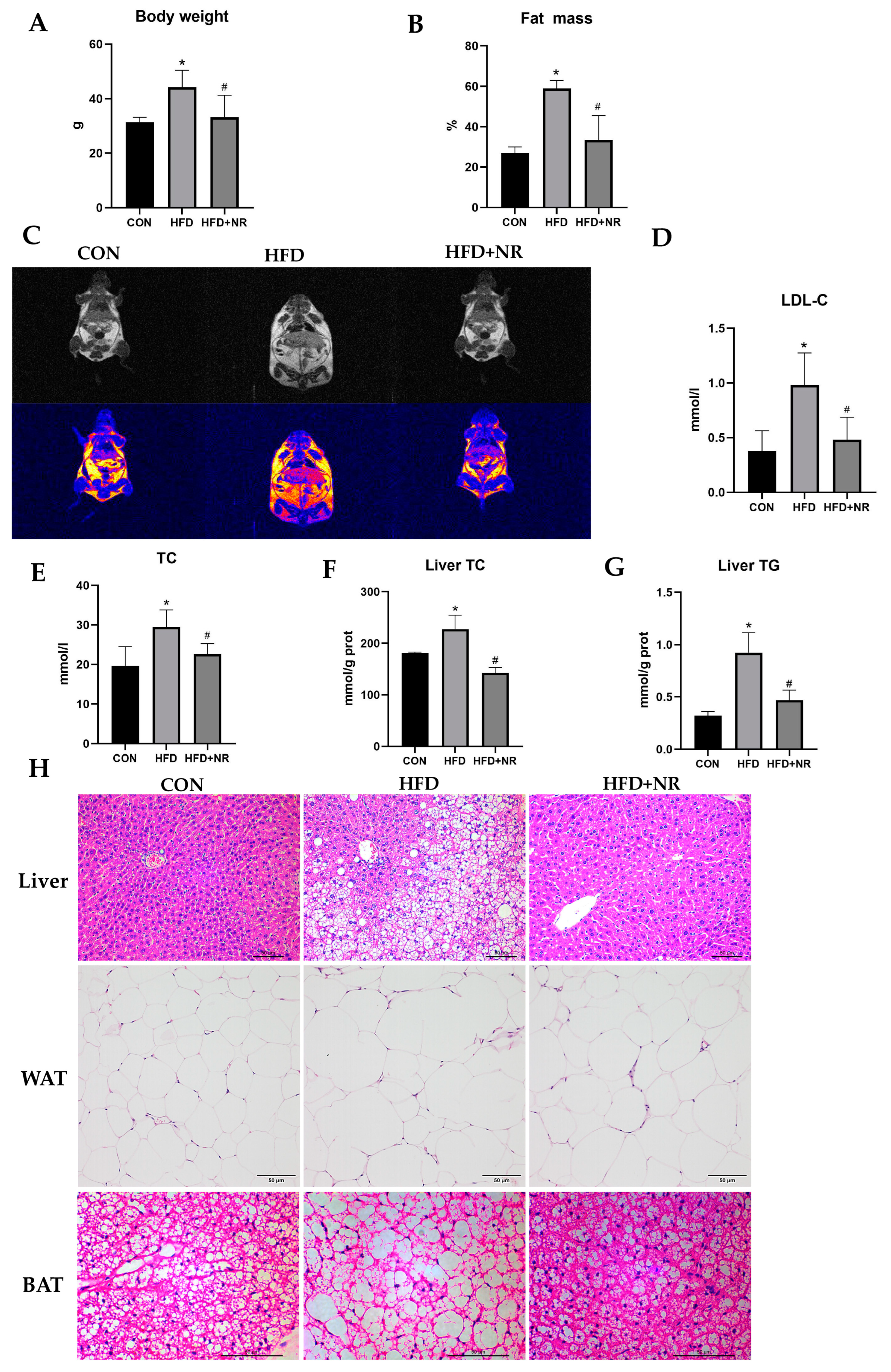
2.1. NR Supplementation Prevents Obesity-Related Metabolic Abnormalities in HFD-Fed Mice
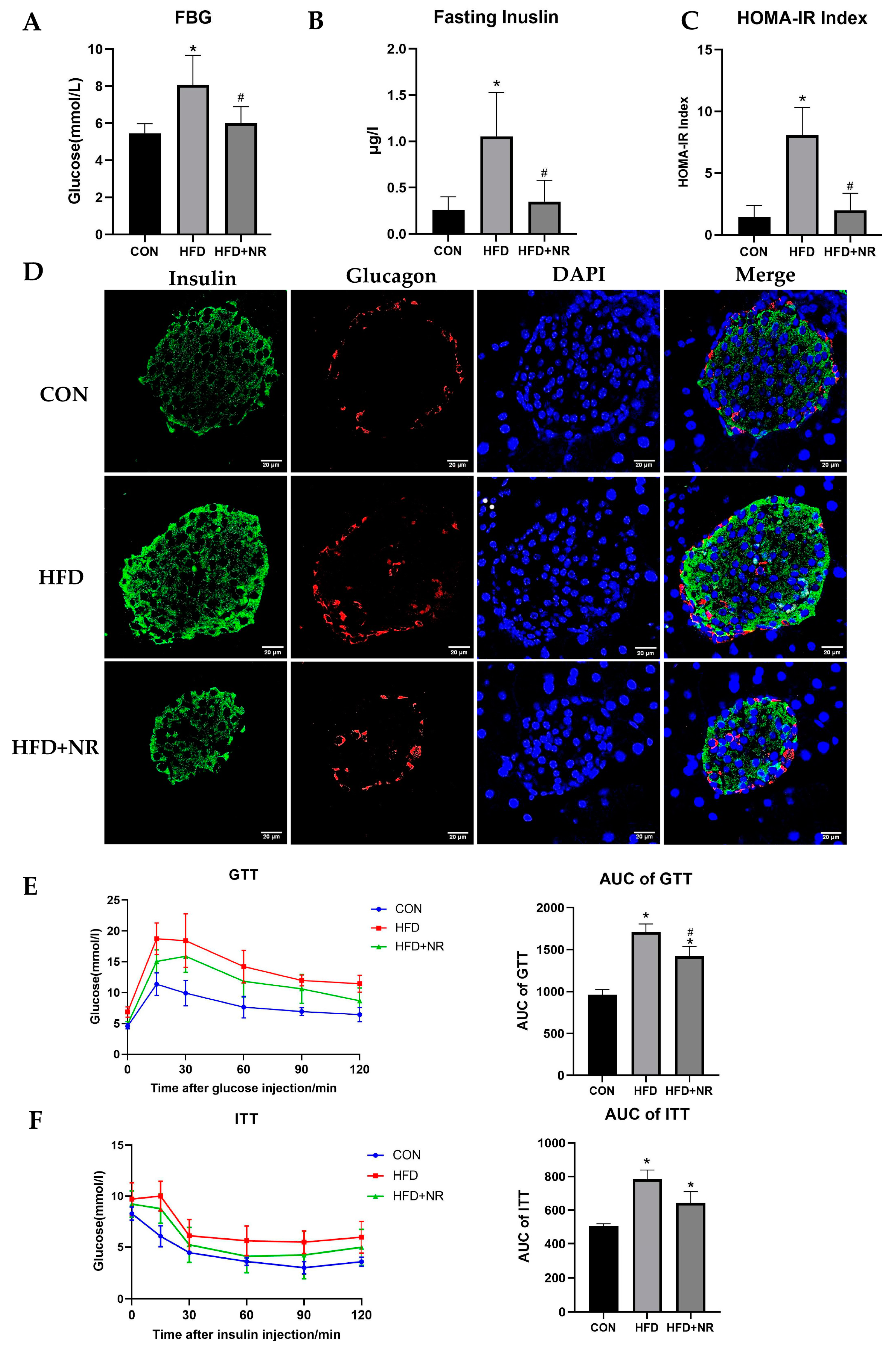
2.2. NR Supplementation Alleviates Insulin Resistance in HFD-Fed Mice
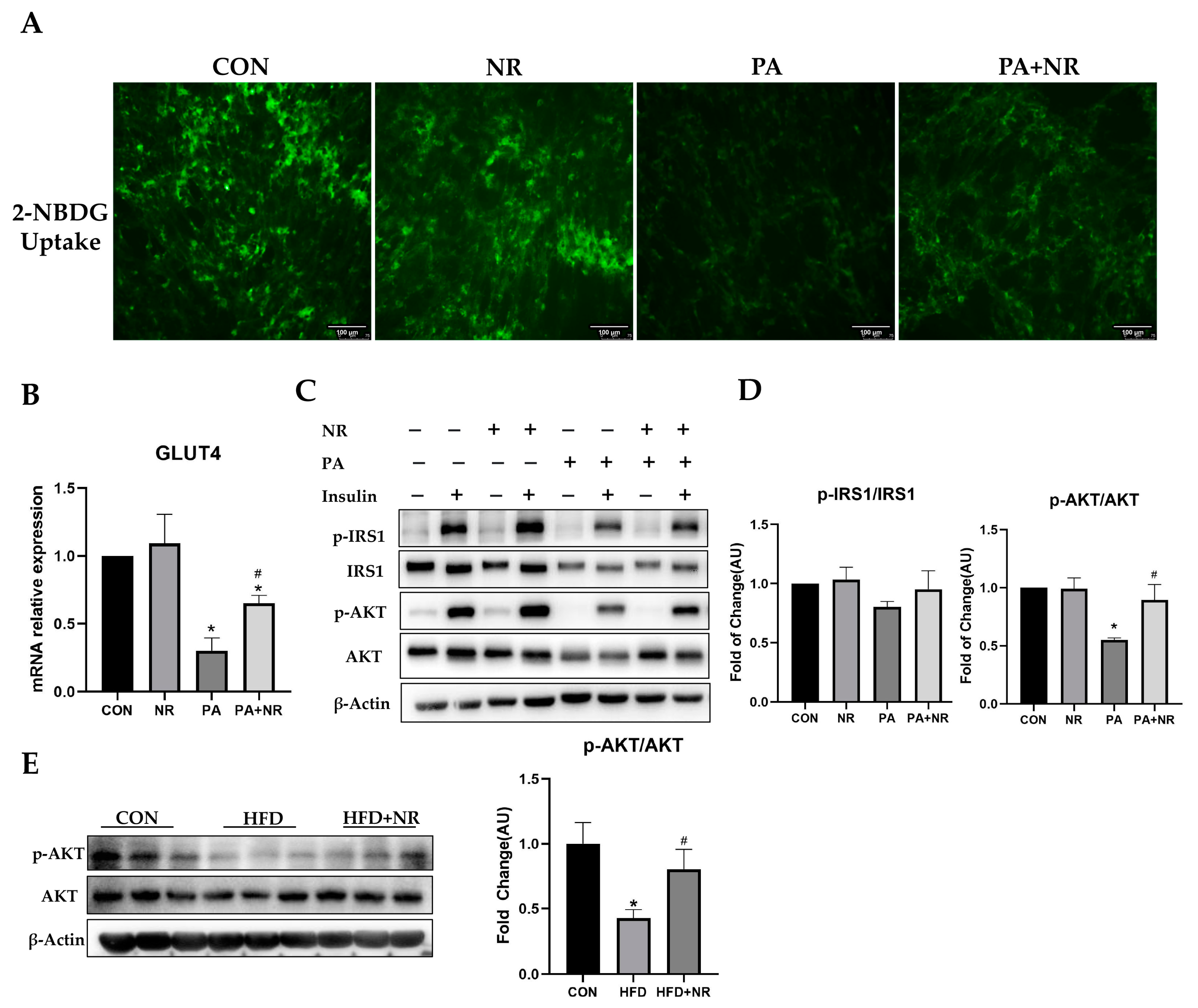
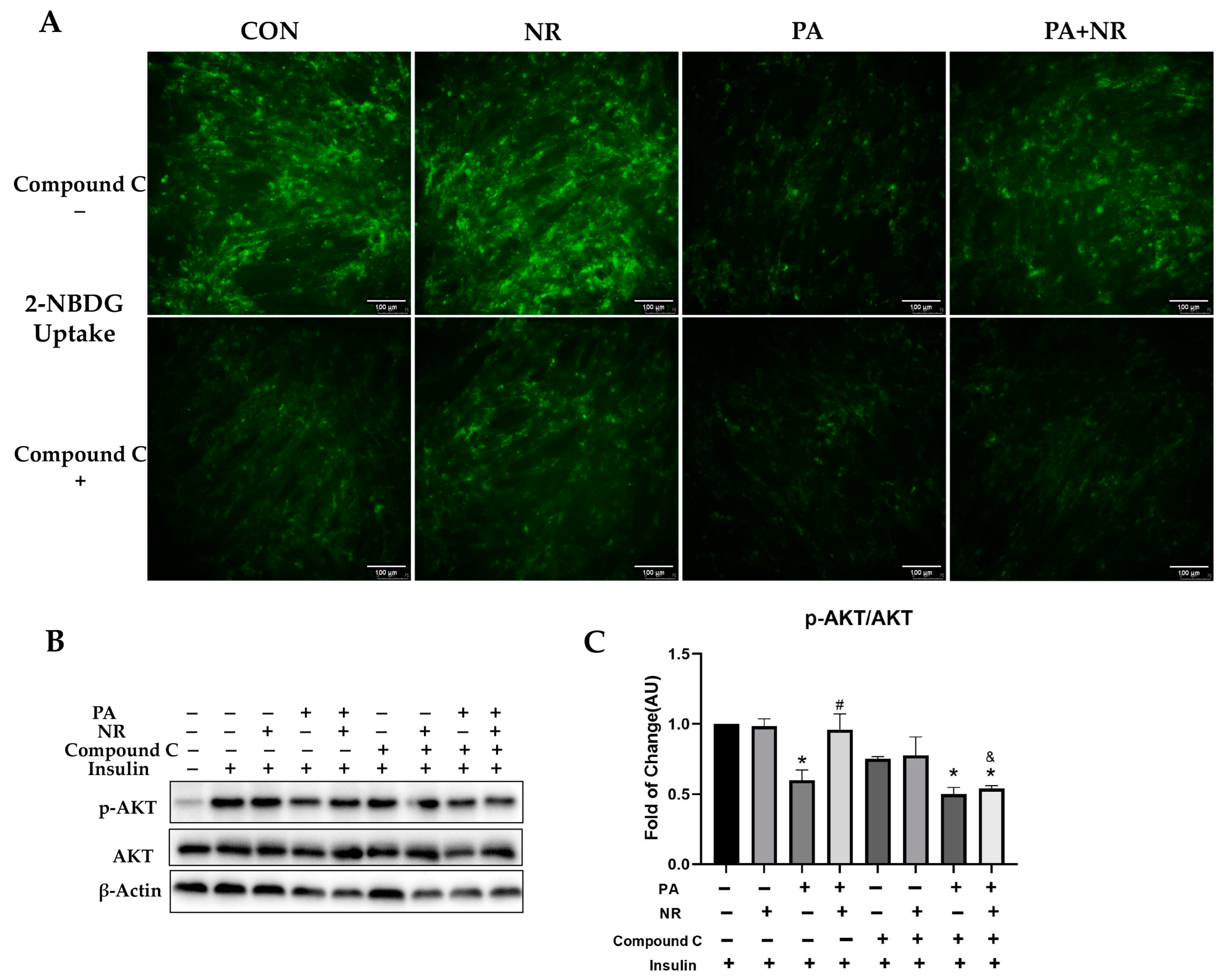
2.3. NR Supplementation Improves Insulin Sensitivity in Skeletal Muscle Cells
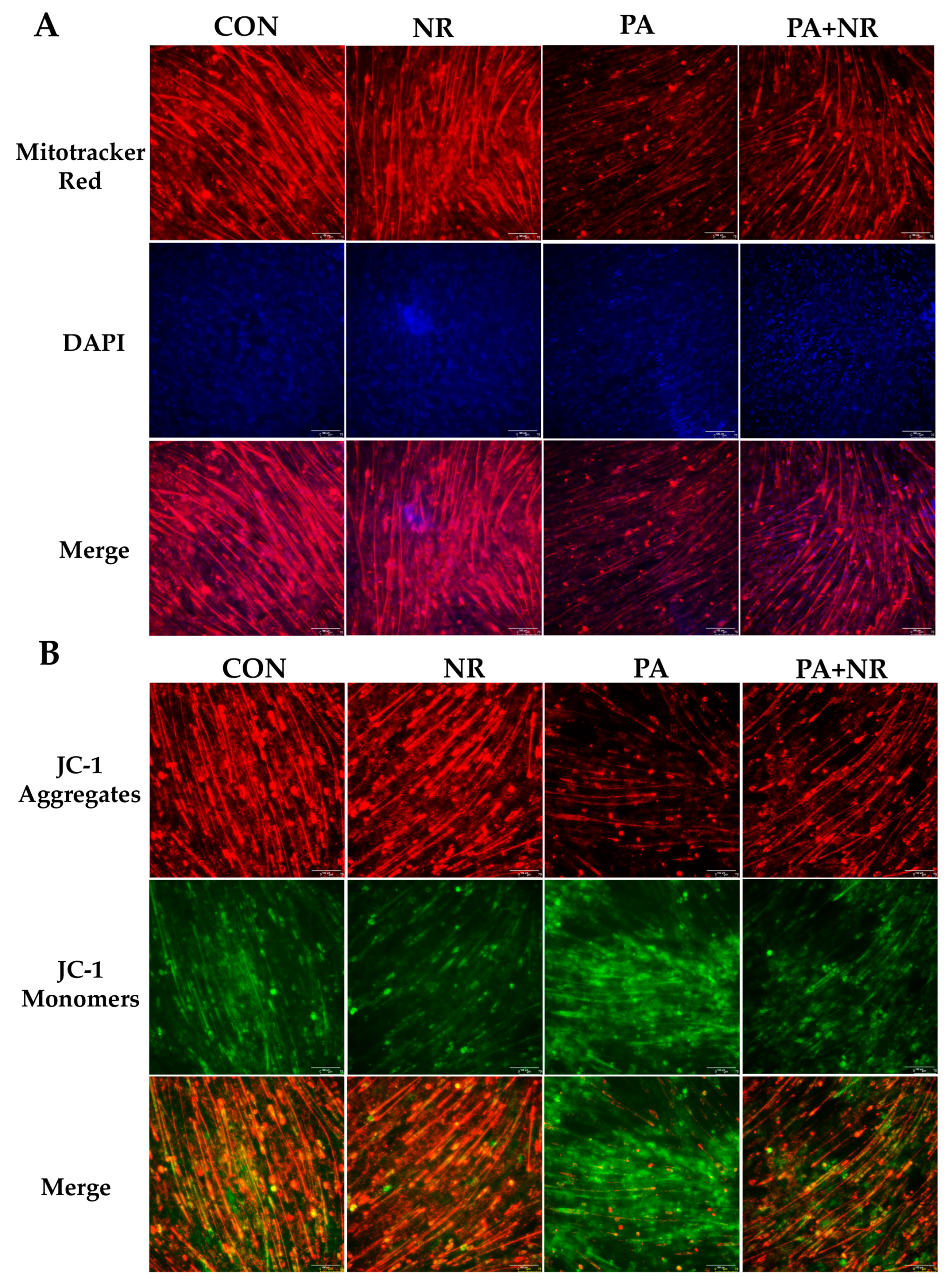
2.4. NR Supplementation Alleviates Mitochondrial Dysfunction Induced by HFD
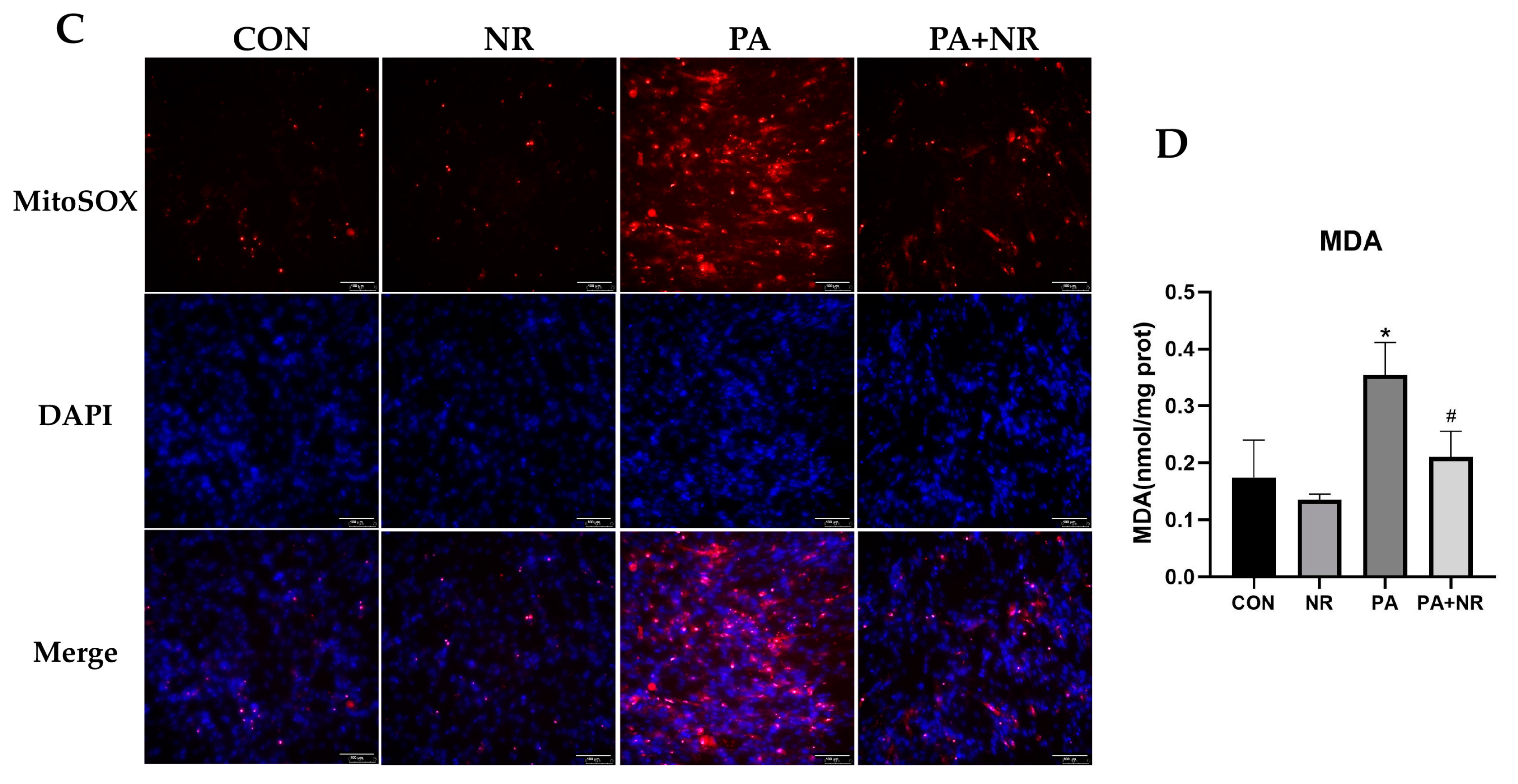
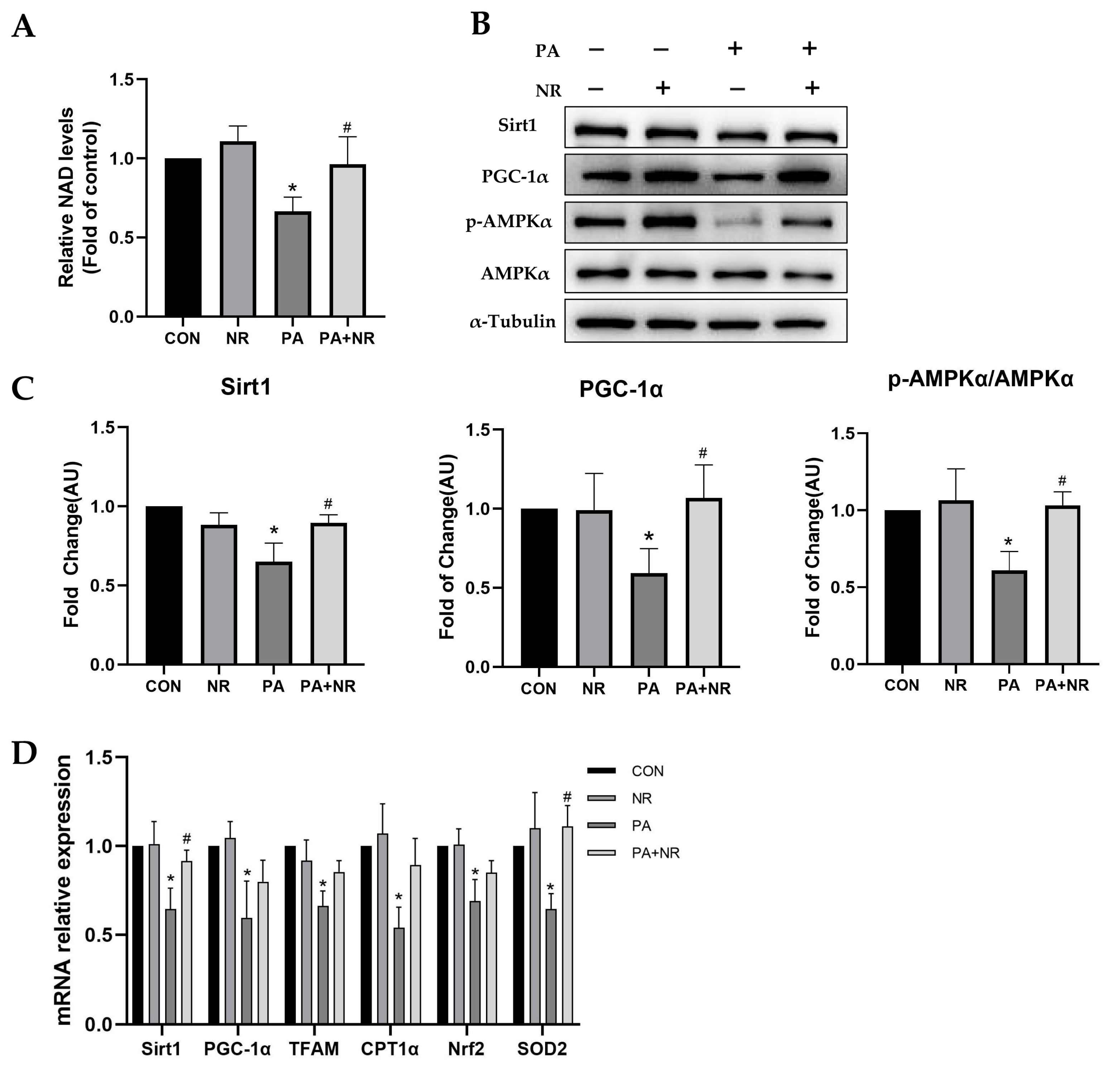
2.5. NR Supplementation Ameliorates Mitochondrial Dysfunction and Oxidative Stress in Skeletal Muscle Cells through Activating AMPK
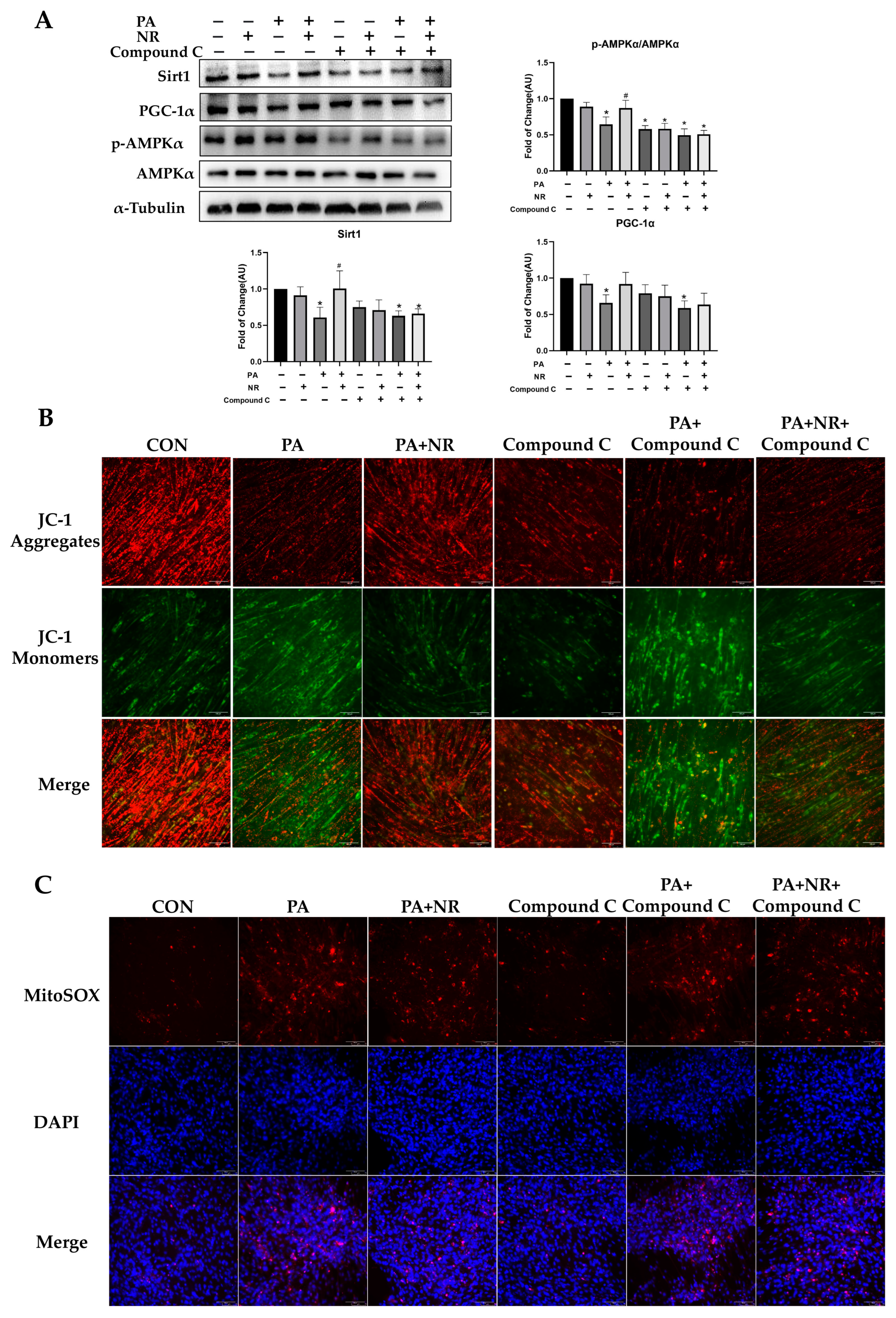
2.6. AMPK Activation Is Required for Improvement on Mitochondrial Function and Insulin Sensitivity via NR in PA-Treated Skeletal Muscle Cells
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.1.1. Intraperitoneal Glucose Tolerance Test (IPGTT) and Insulin Tolerance Test (ITT)
4.1.2. Immunohistochemistry
4.1.3. Immunofluorescence
4.2. Cell Culture Experiments
4.2.1. Insulin-Stimulated Glucose Uptake Analysis
4.2.2. MitoTracker Red Staining
4.2.3. Mitochondrial Membrane Potential Determination
4.2.4. MitoSox Staining
4.2.5. Measurement of MDA Levels
4.2.6. Measurement of NAD Levels
4.2.7. Western Blot Analysis
4.2.8. Quantitative PCR
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IDF. Diabetes Atlas 2021, 10th ed.; IDF: Brussels, Belgium, 2021; Available online: https://diabetesatlas.org/data/en/ (accessed on 12 September 2021).
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalyfa, A.; Wang, Y.; Zhang, S.X.; Qiao, Z.; Abdelkarim, A.; Gozal, D. Sleep Fragmentation in Mice Induces Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2-Dependent Mobilization, Proliferation, and Differentiation of Adipocyte Progenitors in Visceral White Adipose Tissue. Sleep 2014, 37, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Chomentowski, P.; Coen, P.; Radiková, Z.; Goodpaster, B.H.; Toledo, F.G.S. Skeletal Muscle Mitochondria in Insulin Resistance: Differences in Intermyofibrillar Versus Subsarcolemmal Subpopulations and Relationship to Metabolic Flexibility. J. Clin. Endocrinol. Metab. 2011, 96, 494–503. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Treebak, J.T.; Frøsig, C.; Pehmøller, C.; Chen, S.; Maarbjerg, S.J.; Brandt, N.; MacKintosh, C.; Zierath, J.R.; Hardie, D.G.; Kiens, B.; et al. Potential role of TBC1D4 in enhanced post-exercise insulin action in human skeletal muscle. Diabetologia 2009, 52, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wasserman, D.H.; MacKintosh, C.; Sakamoto, K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 2011, 13, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Pehmøller, C.; Treebak, J.T.; Birk, J.B.; Chen, S.; MacKintosh, C.; Hardie, D.G.; Richter, E.A.; Wojtaszewski, J.F.P.; Hargreaves, M.; Szekeres, F.; et al. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E665–E675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.W.; Hwang, H.-J.; Hong, H.C.; Yoo, H.J.; Baik, S.H.; Choi, K.M. BAIBA attenuates insulin resistance and inflammation induced by palmitate or a high fat diet via an AMPK–PPARδ-dependent pathway in mice. Diabetologia 2015, 58, 2096–2105. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Yoshino, M.; Yoshino, J.; Kayser, B.D.; Patti, G.J.; Franczyk, M.P.; Mills, K.F.; Sindelar, M.; Pietka, T.; Patterson, B.W.; Imai, S.-I.; et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 2021, 372, 1224–1229. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.-I. Nicotinamide Mononucleotide, a Key NAD(+) Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, M.; Nascimento-Ferreira, I.; Orsomando, G.; Mori, V.; Gilley, J.; Brown, R.; Janeckova, L.; Vargas, M.E.; Worrell, L.A.; Loreto, A.; et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015, 22, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Minto, C.; Vecchio, M.G.; Lamprecht, M.; Gregori, D. Definition of a tolerable upper intake level of niacin: A systematic review and meta-analysis of the dose-dependent effects of nicotinamide and nicotinic acid supplementation. Nutr. Rev. 2017, 75, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.D.; Liu, P.; Kamanna, V.S.; Kashyap, M.L. Nicotinic acid induces secretion of prostaglandin D2 in human macrophages: An in vitro model of the niacin flush. Atherosclerosis 2007, 192, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Brakedal, B.; Dölle, C.; Riemer, F.; Ma, Y.; Nido, G.S.; Skeie, G.O.; Craven, A.R.; Schwarzlmüller, T.; Brekke, N.; Diab, J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022, 34, 396–407e6. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Chubanava, S.; Agerholm, M.; Søndergård, S.D.; Altıntaş, A.; Møller, A.B.; Høyer, K.F.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Lavery, G.G.; et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 2020, 598, 731–754. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Chen, Y.; Wang, J.; Hao, L.; Huang, C.; Griffiths, A.; Sun, Z.; Zhou, Z.; Song, Z. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J. Hepatol. 2020, 73, 783–793. [Google Scholar] [CrossRef]
- Wang, Z.H.; Bao, X.G.; Hu, J.J.; Shen, S.B.; Xu, G.H.; Wu, Y.L. Nicotinamide Riboside Enhances Endothelial Precursor Cell Function to Promote Refractory Wound Healing Through Mediating the Sirt1/AMPK Pathway. Front. Pharmacol. 2021, 12, 671563. [Google Scholar] [CrossRef]
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1α/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Zhou, Y.; Wang, S.; Pang, N.; Huang, Y.; Ye, M.; Wan, T.; Qiu, Y.; Pei, L.; Jiang, X.; et al. Nicotinamide riboside protects against liver fibrosis induced by CCl(4) via regulating the acetylation of Smads signaling pathway. Life Sci. 2019, 225, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Stromsdorfer, K.L.; Yamaguchi, S.; Yoon, M.J.; Moseley, A.C.; Franczyk, M.P.; Kelly, S.C.; Qi, N.; Imai, S.-I.; Yoshino, J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016, 16, 1851–1860. [Google Scholar] [CrossRef] [Green Version]
- Li, D.J.; Sun, S.J.; Fu, J.T.; Ouyang, S.X.; Zhao, Q.J.; Su, L.; Ji, Q.X.; Sun, D.Y.; Zhu, J.H.; Zhang, G.Y.; et al. NAD(+)-boosting therapy alleviates nonalcoholic fatty liver disease via stimulating a novel exerkine Fndc5/irisin. Theranostics 2021, 11, 4381–4402. [Google Scholar] [CrossRef] [PubMed]
- De Castro, J.M.; Assumpção, J.A.F.; Stein, D.J.; Toledo, R.S.; da Silva, L.S.; Caumo, W.; Carraro, C.C.; da Rosa Araujo, A.S.; Torres, I.L. Nicotinamide riboside reduces cardiometabolic risk factors and modulates cardiac oxidative stress in obese Wistar rats under caloric restriction. Life Sci. 2020, 263, 118596. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; Van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, R.W.; Guan, H.-P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Trotter, D.G.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Yano, N.; Zhang, L.; Wei, D.; Dubielecka, P.M.; Wei, L.; Zhuang, S.; Zhu, P.; Qin, G.; Liu, P.Y.; Chin, Y.E.; et al. Irisin counteracts high glucose and fatty acid-induced cytotoxicity by preserving the AMPK-insulin receptor signaling axis in C2C12 myoblasts. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E791–E805. [Google Scholar] [CrossRef]
- Gordon, S.E.; Lake, J.A.; Westerkamp, C.M.; Thomson, D.M. Does AMP-activated protein kinase negatively mediate aged fast-twitch skeletal muscle mass? Exerc. Sport Sci. Rev. 2008, 36, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Quattrocelli, M.; Wintzinger, M.; Miz, K.; Levine, D.C.; Peek, C.B.; Bass, J.; McNally, E.M. Muscle mitochondrial remodeling by intermittent glucocorticoid drugs requires an intact circadian clock and muscle PGC1α. Sci. Adv. 2022, 8, eabm1189. [Google Scholar] [CrossRef]
- Befroy, D.E.; Petersen, K.F.; Dufour, S.; Mason, G.F.; de Graaf, R.A.; Rothman, D.L.; Shulman, G.I. Impaired Mitochondrial Substrate Oxidation in Muscle of Insulin-Resistant Offspring of Type 2 Diabetic Patients. Diabetes 2007, 56, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of Subsarcolemmal Mitochondria in Obesity and Type 2 Diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Coll, T.; Eyre, E.; Rodríguez-Calvo, R.; Palomer, X.; Sánchez, E.E.; Merlos, M.; Laguna, J.C.; Vázquez-Carrera, M. Oleate Reverses Palmitate-induced Insulin Resistance and Inflammation in Skeletal Muscle Cells. J. Biol. Chem. 2008, 283, 11107–11116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Davuluri, G.; Fujioka, H.; Dantas, W.S.; Huang, E.; Pergola, K.; Mey, J.T.; King, W.T.; et al. Lipids activate skeletal muscle mitochondrial fission and quality control networks to induce insulin resistance in humans. Metabolism 2021, 121, 154803. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Handschin, C.; Arlow, D.; Xie, X.; St, P.J.; Sihag, S. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. USA 2004, 101, 6570–6575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Nawaz, A.; Kado, T.; Takikawa, A.; Igarashi, Y.; Onogi, Y.; Wada, T.; Sasaoka, T.; Yamamoto, S.; Sasahara, M.; et al. Astaxanthin stimulates mitochondrial biogenesis in insulin resistant muscle via activation of AMPK pathway. J. Cachexia- Sarcopenia Muscle 2020, 11, 241–258. [Google Scholar] [CrossRef] [Green Version]
- Pagel-Langenickel, I.; Bao, J.; Pang, L.; Sack, M.N. The Role of Mitochondria in the Pathophysiology of Skeletal Muscle Insulin Resistance. Endocrinol. Rev. 2010, 31, 25–51. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.; et al. Lower Intrinsic ADP-Stimulated Mitochondrial Respiration Underlies In Vivo Mitochondrial Dysfunction in Muscle of Male Type 2 Diabetic Patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hey-Mogensen, M.; Højlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernström, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Ghani, M.A.; Jani, R.; Chavez, A.; Molina-Carrion, M.; Tripathy, D.; DeFronzo, R.A. Mitochondrial reactive oxygen species generation in obese non-diabetic and type 2 diabetic participants. Diabetologia 2009, 52, 574–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, J.M.; Grimsrud, P.A.; Wright, W.S.; Xu, X.; Foncea, R.E.; Graham, D.W.; Brestoff, J.R.; Wiczer, B.M.; Ilkayeva, O.; Cianflone, K.; et al. Downregulation of Adipose Glutathione S-Transferase A4 Leads to Increased Protein Carbonylation, Oxidative Stress, and Mitochondrial Dysfunction. Diabetes 2010, 59, 1132–1142. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.-N.; Woo, C.-H.; Kim, H.-Y.; Park, S.-Y. Methionine sulfoxide reductase B3 deficiency inhibits the development of diet-induced insulin resistance in mice. Redox Biol. 2021, 38, 101823. [Google Scholar] [CrossRef]
- Lee, H.; Ha, T.Y.; Jung, C.H.; Nirmala, F.S.; Park, S.; Huh, Y.H.; Ahn, J. Mitochondrial dysfunction in skeletal muscle contributes to the development of acute insulin resistance in mice. J. Cachexia- Sarcopenia Muscle 2021, 12, 1925–1939. [Google Scholar] [CrossRef]
- Kim, A.; Koo, J.H.; Jin, X.; Kim, W.; Park, S.; Park, S.; Rhee, E.P.; Choi, C.S.; Kim, S.G. Ablation of USP21 in skeletal muscle promotes oxidative fibre phenotype, inhibiting obesity and type 2 diabetes. J. Cachexia- Sarcopenia Muscle 2021, 12, 1669–1689. [Google Scholar] [CrossRef]
- Carter, C.S.; Huang, S.C.; Searby, C.C.; Cassaidy, B.; Miller, M.J.; Grzesik, W.J.; Piorczynski, T.B.; Pak, T.K.; Walsh, S.A.; Acevedo, M.; et al. Exposure to Static Magnetic and Electric Fields Treats Type 2 Diabetes. Cell Metab. 2020, 32, 561–574.e7. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, J.H.; Zhang, H.; Canfran-Duque, A.; Singh, A.K.; Perry, R.J.; Shulman, G.I.; Fernandez-Hernando, C.; Min, W. Brown adipose TRX2 deficiency activates mtDNA-NLRP3 to impair thermogenesis and protect against diet-induced insulin resistance. J. Clin. Investig. 2022, 132, 148852. [Google Scholar] [CrossRef]
- Liu, W.; Zhao, Y.; Wang, G.; Feng, S.; Ge, X.; Ye, W.; Wang, Z.; Zhu, Y.; Cai, W.; Bai, J.; et al. TRIM22 inhibits osteosarcoma progression through destabilizing NRF2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. 2022, 53, 102344. [Google Scholar] [CrossRef]
- Xiong, W.; Garfinkel, A.E.M.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Chen, Z.; Sun, C.; Yang, D.; Zhou, Z.; Peng, X.; Zheng, L.; Tang, C. Exercise Intervention Mitigates Pathological Liver Changes in NAFLD Zebrafish by Activating SIRT1/AMPK/NRF2 Signaling. Int. J. Mol. Sci. 2021, 22, 10940. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ouyang, Y.; Yin, H.; Cui, H.; Deng, H.; Liu, H.; Jian, Z.; Fang, J.; Zuo, Z.; Wang, X.; et al. Induction of autophagy via the ROS-dependent AMPK-mTOR pathway protects copper-induced spermatogenesis disorder. Redox Biol. 2022, 49, 102227. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primers | Sequences (Primer: 5′-3′) |
|---|---|---|
| SOD2 | Forward primer | GCCCAAACCTATCGTGTCCA |
| Reverse primer | AGGGAACCCTAAATGCTGCC | |
| β-Actin | Forward primer | GTGGTGGTGAAGCTGTAGCC |
| Reverse primer | AGCCATGTACGTAGCCATCC | |
| Sirt1 | Forward primer | TGTGAAGTTACTGCAGGAGTGTAAA |
| Reverse primer | GCATAGATACCGTCTCTTGATCTGAA | |
| PGC-1α | Forward primer | AAGTGTGGAACTCTCTGGAACTG |
| Reverse primer | GGGTTATCTTGGTTGGCTTTATG | |
| TFAM | Forward primer | AACACCCAGATGCAAAACTTTCA |
| Reverse primer | GACTTGGAGTTAGCTGCTCTTT | |
| Nrf2 | Forward primer | CTTTAGTCAGCGACAGAAGGAC |
| Reverse primer | AGGCATCTTGTTTGGGAATGTG | |
| CPT-1α | Forward primer | CTCCGCCTGAGCCATGAAG |
| Reverse primer | CACCAGTGATGATGCCATTCT | |
| GLUT4 | Forward primer | GTGACTGGAACACTGGTCCTA |
| Reverse primer | CCAGCCACGTTGCATTGTAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Jiang, X.; Zhou, Y.; Gu, Y.; Ding, Y.; Luo, J.; Pang, N.; Sun, Y.; Pei, L.; Pan, J.; et al. Improving Mitochondrial Function in Skeletal Muscle Contributes to the Amelioration of Insulin Resistance by Nicotinamide Riboside. Int. J. Mol. Sci. 2023, 24, 10015. https://doi.org/10.3390/ijms241210015
Li Q, Jiang X, Zhou Y, Gu Y, Ding Y, Luo J, Pang N, Sun Y, Pei L, Pan J, et al. Improving Mitochondrial Function in Skeletal Muscle Contributes to the Amelioration of Insulin Resistance by Nicotinamide Riboside. International Journal of Molecular Sciences. 2023; 24(12):10015. https://doi.org/10.3390/ijms241210015
Chicago/Turabian StyleLi, Qiuyan, Xuye Jiang, Yujia Zhou, Yingying Gu, Yijie Ding, Jing Luo, Nengzhi Pang, Yan Sun, Lei Pei, Jie Pan, and et al. 2023. "Improving Mitochondrial Function in Skeletal Muscle Contributes to the Amelioration of Insulin Resistance by Nicotinamide Riboside" International Journal of Molecular Sciences 24, no. 12: 10015. https://doi.org/10.3390/ijms241210015
APA StyleLi, Q., Jiang, X., Zhou, Y., Gu, Y., Ding, Y., Luo, J., Pang, N., Sun, Y., Pei, L., Pan, J., Gao, M., Ma, S., Xiao, Y., Hu, D., Wu, F., & Yang, L. (2023). Improving Mitochondrial Function in Skeletal Muscle Contributes to the Amelioration of Insulin Resistance by Nicotinamide Riboside. International Journal of Molecular Sciences, 24(12), 10015. https://doi.org/10.3390/ijms241210015