
IGF-1 Stimulates Glycolytic ATP Production in MCF-7L Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The IGF System Structure and Signaling
1.2. IGF Axis in Metabolism and Breast Cancer Risk
1.3. Targeting the IGF System
2. Results
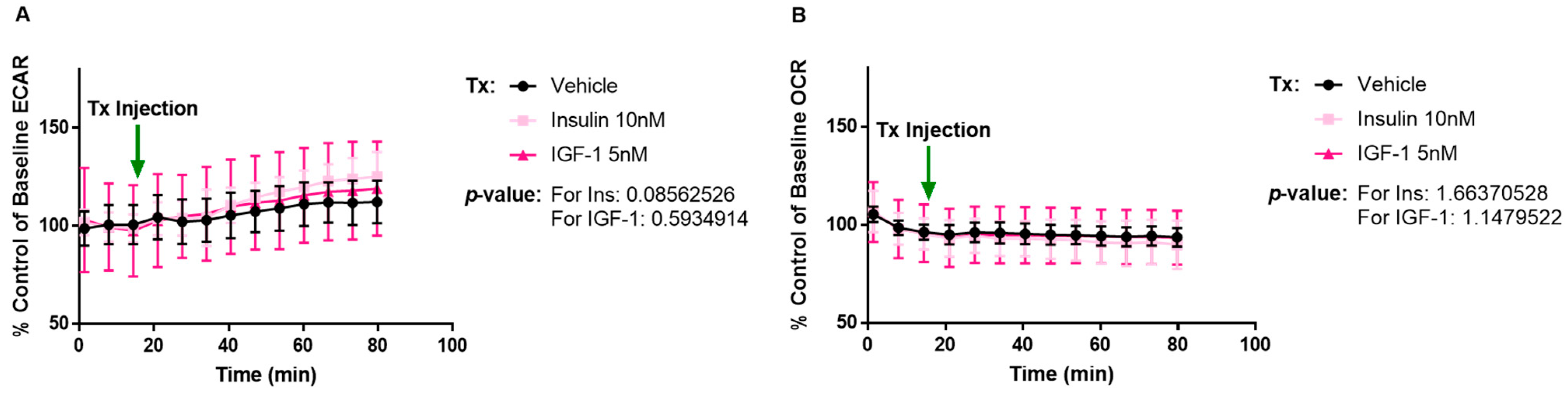
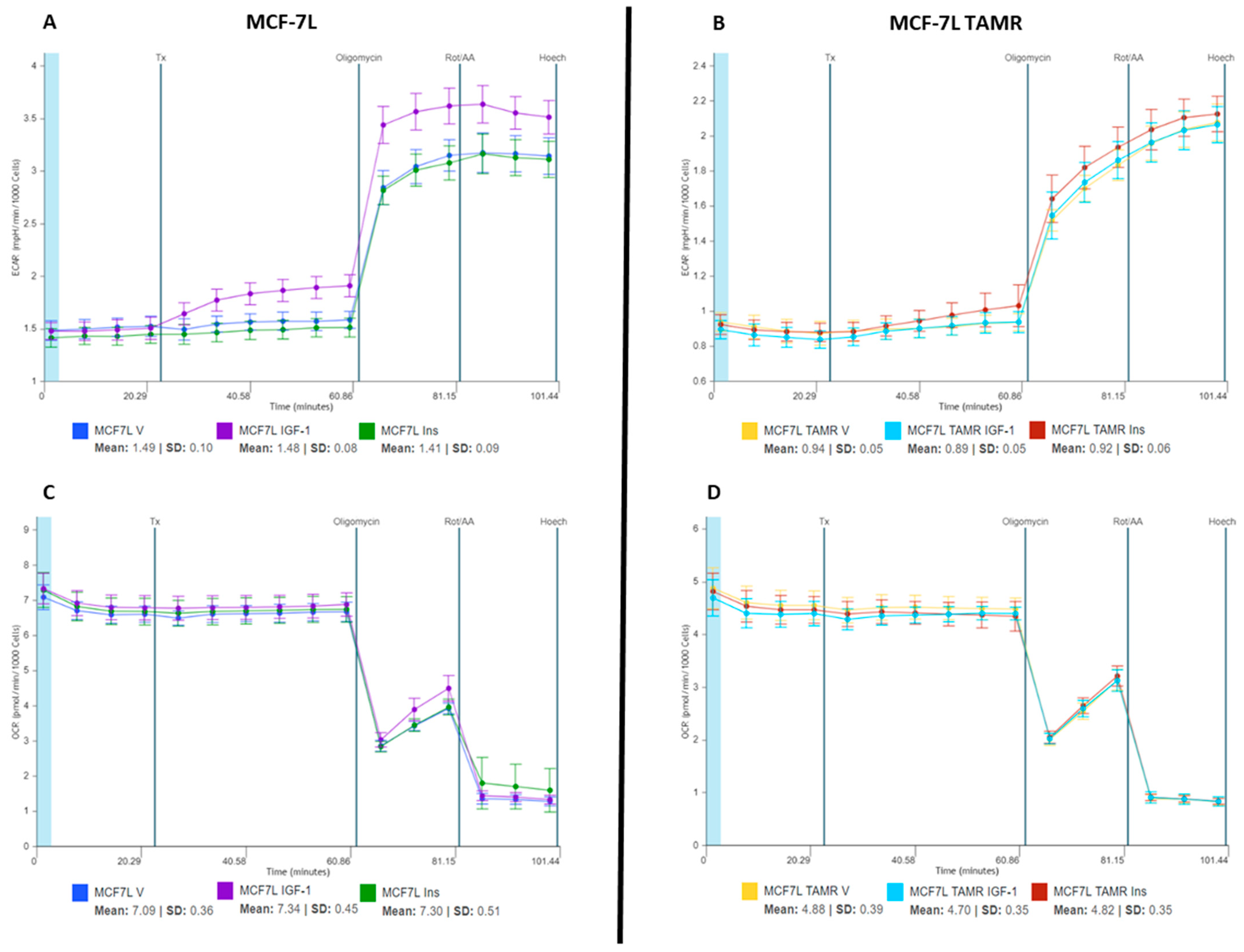
2.1. IGF-1 Increases ECAR Response of MCF-7L Cells
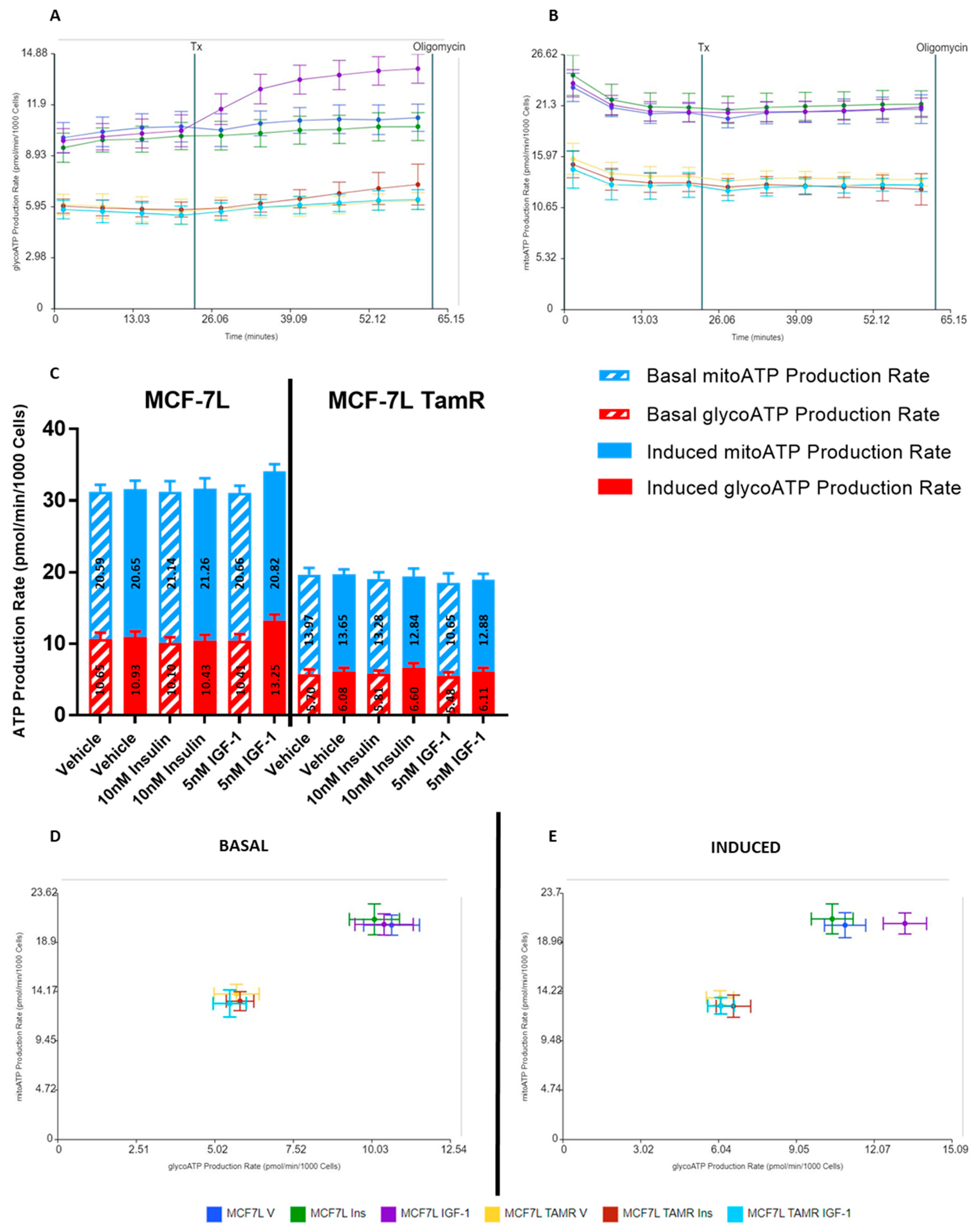
2.2. IGF-1 Upregulates Glycolysis in MCF-7L Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell and Cell Culture
4.3. Mitochondrial Bioenergetics
4.4. Calculations
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breast Cancer Statistics|How Common Is Breast Cancer? Available online: https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html (accessed on 25 January 2023).
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T. Molecular and Cellular Aspects of the Insulin-like Growth Factor I Receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.S.; Rogol, A.D.; Rosenfeld, R.G. The History of the Insulin-Like Growth Factor System. Horm. Res. Paediatr. 2022, 95, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Yerushalmi, R.; Gelmon, K.A.; Leung, S.; Gao, D.; Cheang, M.; Pollak, M.; Turashvili, G.; Gilks, B.C.; Kennecke, H. Insulin-like Growth Factor Receptor (IGF-1R) in Breast Cancer Subtypes. Breast Cancer Res. Treat. 2012, 132, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; LeRoith, D. Hallmarks of Cancer: The Insulin-like Growth Factors Perspective. Front. Oncol. 2022, 12, 1055589. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, Insulin, and Cancer. Endocrinology 2011, 152, 2546–2551. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M. The Insulin and Insulin-like Growth Factor Receptor Family in Neoplasia: An Update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- LeRoith, D.; Yakar, S. Mechanisms of Disease: Metabolic Effects of Growth Hormone and Insulin-like Growth Factor 1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef]
- Cai, W.; Sakaguchi, M.; Kleinridders, A.; Gonzalez-Del Pino, G.; Dreyfuss, J.M.; O’Neill, B.T.; Ramirez, A.K.; Pan, H.; Winnay, J.N.; Boucher, J.; et al. Domain-Dependent Effects of Insulin and IGF-1 Receptors on Signalling and Gene Expression. Nat. Commun. 2017, 8, 14892. [Google Scholar] [CrossRef] [Green Version]
- De Meyts, P.; Whittaker, J. Structural Biology of Insulin and IGF1 Receptors: Implications for Drug Design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef]
- De Meyts, P. Insulin and Its Receptor: Structure, Function and Evolution. BioEssays 2004, 26, 1351–1362. [Google Scholar] [CrossRef]
- Meyts, P.D.; Wallach, B.; Christoffersen, C.T.; Ursø, B.; Grønskov, K.; Latus, L.-J.; Yakushiji, F.; Ilondo, M.; Shymko, R.M. The Insulin-Like Growth Factor-I Receptor. Horm. Res. Paediatr. 1994, 42, 152–169. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.-I. 40 YEARS OF IGF1: IGF1 Receptor Signaling Pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Flier, J.S.; Benecke, H.; Ransil, B.J.; Moller, D.E. Ligand-Binding Properties of the Two Isoforms of the Human Insulin Receptor. Endocrinology 1993, 132, 1132–1138. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Characterization of Insulin/IGF Hybrid Receptors: Contributions of the Insulin Receptor L2 and Fn1 Domains and the Alternatively Spliced Exon 11 Sequence to Ligand Binding and Receptor Activation|Biochemical Journal|Portland Press. Available online: https://portlandpress-com.ezp2.lib.umn.edu/biochemj/article/403/3/603/42434/Characterization-of-insulin-IGF-hybrid-receptors (accessed on 13 April 2023).
- Yang, Y.; Yee, D. Targeting Insulin and Insulin-Like Growth Factor Signaling in Breast Cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef]
- Cao, J.; Yee, D. Disrupting Insulin and IGF Receptor Function in Cancer. Int. J. Mol. Sci. 2021, 22, 555. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, X.; Wang, Y.; Sun, G.; Zhang, J.; Li, Z. Obesity and Endocrine-Related Cancer: The Important Role of IGF-1. Front. Endocrinol. 2023, 14, 1093257. [Google Scholar] [CrossRef]
- Guler, H.P.; Zapf, J.; Froesch, E.R. Short-Term Metabolic Effects of Recombinant Human Insulin-like Growth Factor I in Healthy Adults. N. Engl. J. Med. 1987, 317, 137–140. [Google Scholar] [CrossRef]
- Jacob, R.; Barrett, E.; Plewe, G.; Fagin, K.D.; Sherwin, R.S. Acute Effects of Insulin-like Growth Factor I on Glucose and Amino Acid Metabolism in the Awake Fasted Rat. Comparison with Insulin. J. Clin. Investig. 1989, 83, 1717–1723. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Tocheny, C.E.; Shaw, L.M. The Insulin-like Growth Factor Signaling Pathway in Breast Cancer: An Elusive Therapeutic Target. Life 2022, 12, 1992. [Google Scholar] [CrossRef]
- Insulin-Like Growth Factor-II: Its Role in Metabolic and Endocrine Disease—Livingstone—2014—Clinical Endocrinology—Wiley Online Library. Available online: https://onlinelibrary-wiley-com.ezp3.lib.umn.edu/doi/full/10.1111/cen.12446 (accessed on 21 April 2023).
- Biello, F.; Platini, F.; D’Avanzo, F.; Cattrini, C.; Mennitto, A.; Genestroni, S.; Martini, V.; Marzullo, P.; Aimaretti, G.; Gennari, A. Insulin/IGF Axis in Breast Cancer: Clinical Evidence and Translational Insights. Biomolecules 2021, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic Profiling of Triple-Negative Breast Cancer Cells Reveals Metabolic Vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Vella, V.; Nicolosi, M.L.; Giuliano, M.; Morrione, A.; Malaguarnera, R.; Belfiore, A. Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells 2019, 8, 1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. The Metabolism of Carcinoma Cells1. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Zhang, C.; Le, A. Glucose Metabolism in Cancer: The Warburg Effect and Beyond. In The Heterogeneity of Cancer Metabolism; Le, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; pp. 3–15. ISBN 978-3-030-65768-0. [Google Scholar]
- Gatenby, R.A.; Gillies, R.J. Why Do Cancers Have High Aerobic Glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M. Insulin and Insulin-like Growth Factor Signalling in Neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Papa, V.; Pezzino, V.; Costantino, A.; Belfiore, A.; Giuffrida, D.; Frittitta, L.; Vannelli, G.B.; Brand, R.; Goldfine, I.D.; Vigneri, R. Elevated Insulin Receptor Content in Human Breast Cancer. J. Clin. Investig. 1990, 86, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Leroith, D.; Brodt, P. The Role of the Growth Hormone/Insulin-like Growth Factor Axis in Tumor Growth and Progression: Lessons from Animal Models. Cytokine Growth Factor Rev. 2005, 16, 407–420. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Navarro Rosenblatt, D.; Thune, I.; Vieira, R.; Norat, T. Body Mass Index and Survival in Women with Breast Cancer-Systematic Literature Review and Meta-Analysis of 82 Follow-up Studies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 1901–1914. [Google Scholar] [CrossRef]
- Burtrum, D.; Zhu, Z.; Lu, D.; Anderson, D.M.; Prewett, M.; Pereira, D.S.; Bassi, R.; Abdullah, R.; Hooper, A.T.; Koo, H.; et al. A Fully Human Monoclonal Antibody to the Insulin-like Growth Factor I Receptor Blocks Ligand-Dependent Signaling and Inhibits Human Tumor Growth in Vivo. Cancer Res. 2003, 63, 8912–8921. [Google Scholar]
- Beltran, P.J.; Mitchell, P.; Chung, Y.-A.; Cajulis, E.; Lu, J.; Belmontes, B.; Ho, J.; Tsai, M.M.; Zhu, M.; Vonderfecht, S.; et al. AMG 479, a Fully Human Anti–Insulin-like Growth Factor Receptor Type I Monoclonal Antibody, Inhibits the Growth and Survival of Pancreatic Carcinoma Cells. Mol. Cancer Ther. 2009, 8, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Discovery of OSI-906: A Selective and Orally Efficacious Dual Inhibitor of the IGF-1 Receptor and Insulin Receptor|Future Medicinal Chemistry. Available online: https://www.future-science.com/doi/full/10.4155/fmc.09.89 (accessed on 1 April 2023).
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a Small-Molecule Inhibitor of Insulin-like Growth Factor-1 Receptor/Insulin Receptor, Enhances Gemcitabine Response in Pancreatic Cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; LaPara, K.; Yee, D. Disruption of Insulin Receptor Function Inhibits Proliferation in Endocrine-Resistant Breast Cancer Cells. Oncogene 2016, 35, 4235–4243. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.F.; Ferrero, J.-M.; Bourgeois, H.; Kennecke, H.; de Boer, R.H.; Jacot, W.; McGreivy, J.; Suzuki, S.; Zhu, M.; McCaffery, I.; et al. Ganitumab with Either Exemestane or Fulvestrant for Postmenopausal Women with Advanced, Hormone-Receptor-Positive Breast Cancer: A Randomised, Controlled, Double-Blind, Phase 2 Trial. Lancet Oncol. 2013, 14, 228–235. [Google Scholar] [CrossRef]
- Haluska, P.; Shaw, H.M.; Batzel, G.N.; Yin, D.; Molina, J.R.; Molife, L.R.; Yap, T.A.; Roberts, M.L.; Sharma, A.; Gualberto, A.; et al. Phase I Dose Escalation Study of the Anti–Insulin-Like Growth Factor-I Receptor Monoclonal Antibody CP-751,871 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2007, 13, 5834–5840. [Google Scholar] [CrossRef] [Green Version]
- Investigating New Therapeutic Strategies Targeting Hyperinsulinemia’s Mitogenic Effects in a Female Mouse Breast Cancer Model|Endocrinology|Oxford Academic. Available online: https://academic.oup.com/endo/article/154/5/1701/2423371 (accessed on 1 April 2023).
- Forest, A.; Amatulli, M.; Ludwig, D.L.; Damoci, C.B.; Wang, Y.; Burns, C.A.; Donoho, G.P.; Zanella, N.; Fiebig, H.H.; Prewett, M.C.; et al. Intrinsic Resistance to Cixutumumab Is Conferred by Distinct Isoforms of the Insulin Receptor. Mol. Cancer Res. 2015, 13, 1615–1626. [Google Scholar] [CrossRef] [Green Version]
- Fagan, D.H.; Uselman, R.R.; Sachdev, D.; Yee, D. Acquired Resistance to Tamoxifen Is Associated with Loss of the Type I Insulin-like Growth Factor Receptor: Implications for Breast Cancer Treatment. Cancer Res. 2012, 72, 3372–3380. [Google Scholar] [CrossRef] [Green Version]
- Valcarcel-Jimenez, L.; Gaude, E.; Torrano, V.; Frezza, C.; Carracedo, A. Mitochondrial Metabolism: Yin and Yang for Tumor Progression. Trends Endocrinol. Metab. TEM 2017, 28, 748–757. [Google Scholar] [CrossRef]
- Kasprzak, A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Kobayashi, T.; Matsumoto, M.; Watanabe, S.; Yoneda, A.; Bando, Y. Adipose Tissue:Critical Contributor to the Development of Prostate Cancer. J. Med. Investig. JMI 2018, 65, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Palladino, C.; Spoleti, C.; Massimino, M.; Vigneri, P.; Purrello, M.; Ragusa, M.; Morrione, A.; et al. Discoidin Domain Receptor 1 Modulates Insulin Receptor Signaling and Biological Responses in Breast Cancer Cells. Oncotarget 2017, 8, 43248–43270. [Google Scholar] [CrossRef] [Green Version]
- Vella, V.; Milluzzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin Receptor Isoforms in Cancer. Int. J. Mol. Sci. 2018, 19, 3615. [Google Scholar] [CrossRef] [Green Version]
- Gradishar, W.J.; Yardley, D.A.; Layman, R.; Sparano, J.A.; Chuang, E.; Northfelt, D.W.; Schwartz, G.N.; Youssoufian, H.; Tang, S.; Novosiadly, R.; et al. Clinical and Translational Results of a Phase II, Randomized Trial of an Anti-IGF-1R (Cixutumumab) in Women with Breast Cancer That Progressed on Endocrine Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 301–309. [Google Scholar] [CrossRef] [Green Version]
- XF Real-Time ATP Rate Assay Kit User Guide. Available online: https://www.agilent.com/cs/library/usermanuals/public/103592-400_Seahorse_XF_ATP_Rate_kit_User_Guide.pdf (accessed on 16 April 2023).
- GraphPad Prism 9 Statistics Guide—Comparing ROC Curves. Available online: https://www.graphpad.com/guides/prism/latest/statistics/stat_comparing_roc_curves.htm (accessed on 16 April 2023).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajoria, B.; Zhang, X.; Yee, D. IGF-1 Stimulates Glycolytic ATP Production in MCF-7L Cells. Int. J. Mol. Sci. 2023, 24, 10209. https://doi.org/10.3390/ijms241210209
Rajoria B, Zhang X, Yee D. IGF-1 Stimulates Glycolytic ATP Production in MCF-7L Cells. International Journal of Molecular Sciences. 2023; 24(12):10209. https://doi.org/10.3390/ijms241210209
Chicago/Turabian StyleRajoria, Bhumika, Xihong Zhang, and Douglas Yee. 2023. "IGF-1 Stimulates Glycolytic ATP Production in MCF-7L Cells" International Journal of Molecular Sciences 24, no. 12: 10209. https://doi.org/10.3390/ijms241210209