
RNA-Seq Transcriptome Analysis and Evolution of OsEBS, a Gene Involved in Enhanced Spikelet Number per Panicle in Rice

 , ,
, ,
Abstract
:1. Introduction
2. Results
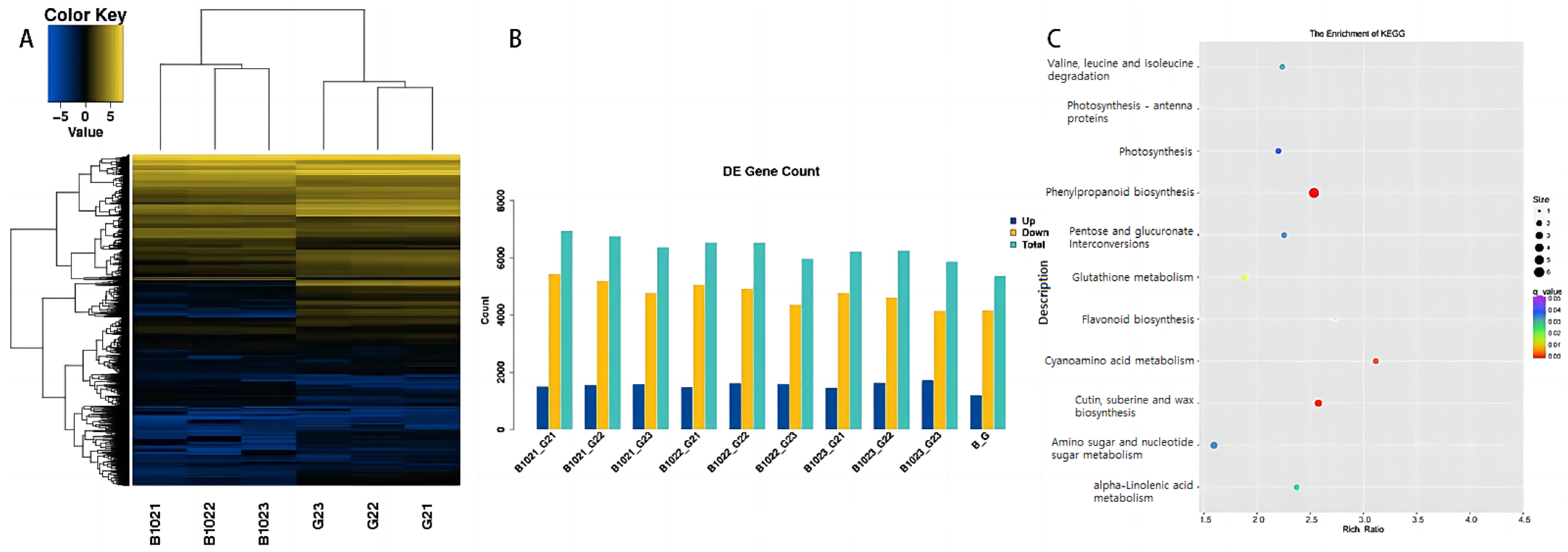
2.1. RNA Sequencing and Sequence Alignment of Guichao 2 and B102
2.2. Analysis of Differentially Expressed Genes (DEGs) in the Panicle of Guichao 2 and B102
2.3. Function Enrichment Analysis
2.4. OsEBS Participated in the Differentiation of Indica and Japonica Subspecies
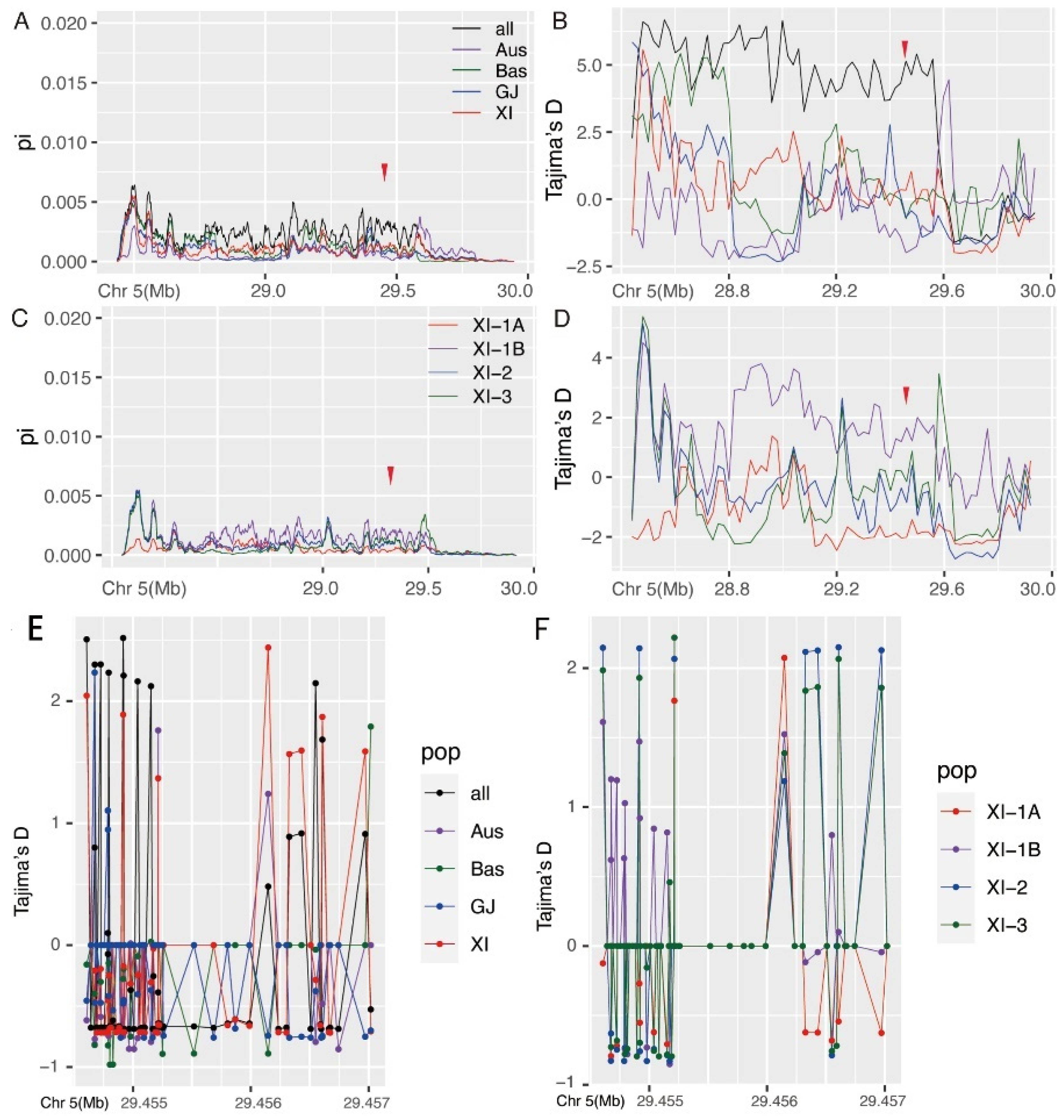
2.5. Population Genetic Analysis
2.6. Accelerated Evolution and Domain Loss of OsEBS Resulted in Neofunctionalization
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growing Conditions
4.2. DNA Extraction and Genotype Identification
4.3. Total RNA Extraction and cDNA Library Construction
4.4. RNA Sequencing (RNA-seq), Data Filtering, and Sequence Alignment
4.5. Gene Differential Expression Analysis
4.6. Function Enrichment Analysis
4.7. Population Genetic Analysis
4.8. Sequence and Phylogenetic Analyses
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Babu, P.D.; Subhasree, R.S.; Bhakyaraj, R. Brown Rice-beyond the color reviving a lost health food—A review. Am.-Eurasian J. Agron. 2009, 2, 67–72. Available online: https://www.researchgate.net/publication/238713802 (accessed on 28 April 2023).
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Makoto, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Kunihiro, S.; Saito, T.; Matsuda, T.; Inoue, M.; Kuramata, M.; Taguchi-Shiobara, F.; Youssefian, S.; Berberich, T.; Kusano, T. Rice DEP1, encoding a highly cysteine-rich G protein γ subunit, confers cadmium tolerance on yeast cells and plants. J. Exp. Bot. 2013, 64, 4517–4527. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Z.; Qian, Q.; Liu, Z.B.; Sun, H.Y.; He, S.Y.; Luo, D.; Xia, G.M.; Chu, C.C.; Li, J.Y.; Fu, X.D. Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.Z.; Liu, Z.X.; Qin, T.; Wang, L.; Chen, Z.H.; Zhang, B.M.; Zhang, H.T.; Li, H.T.; Liu, L.; et al. OsSPL14 acts upstream of OsPIN1b and PILS6b to modulate axillary bud outgrowth by fine-tuning auxin transport in rice. Plant J. 2022, 111, 1167–1182. [Google Scholar] [CrossRef]
- Yuan, H.; Qin, P.; Hu, L.; Zhan, S.J.; Wang, S.F.; Gao, P.; Li, J.; Jin, M.Y.; Xu, Z.Y.; Gao, Q.; et al. OsSPL18 controls grain weight and grain number in rice. J. Genet. Genom. 2019, 46, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.F.; Yu, H.; Xiong, G.S.; Wang, J.; Jiao, Y.Q.; Liu, G.F.; Jing, Y.H.; Meng, X.B.; Hu, X.M.; Qian, Q.; et al. Genome-wide binding analysis of the transcription activator IDEAL PLANT ARCHITECTURE1 reveals a complex network regulating rice plant architecture. Plant Cell 2013, 25, 3743–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.Q.; Wang, Y.H.; Xue, D.W.; Wang, J.; Yan, M.X.; Liu, G.F.; Dong, G.J.; Zeng, D.L.; Lu, Z.F.; Zhu, X.D.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Hua, K.; Xu, R.; Zeng, D.L.; Wang, R.C.; Dong, G.J.; Zhang, G.Z.; Lu, X.L.; Fang, N.; Wang, D.K.; et al. The LARGE2-APO1/APO2 regulatory module controls panicle size and grain number in rice. Plant Cell 2021, 33, 1212–1228. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.Y.; Xing, Y.Z.; Weng, X.Y.; Zhao, Y.; Tang, W.J.; Wang, L.; Zhou, H.J.; Yu, S.B.; Xu, C.G.; Li, X.H.; et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 2008, 40, 761–767. [Google Scholar] [CrossRef]
- Agata, A.; Ando, K.; Ota, S.; Kojima, M.; Takebayashi, Y.; Takehara, S.; Doi, K.; Ueguchi-Tanaka, M.; Suzuki, T.; Sakakibara, H.; et al. Diverse panicle architecture results from various combinations of Prl5/GA20ox4 and Pbl6/APO1 alleles. Commun. Biol. 2020, 3, 302. [Google Scholar] [CrossRef]
- Miao, Y.L.; Xun, Q.; Taji, T.; Keisuke, T.; Yasuno, N.; Ding, C.Q.; Kyozuka, J. ABERRANT PANICLE ORGANIZATION2 controls multiple steps in panicle formation through common direct-target genes. Plant Physiol. 2022, 189, 2210–2226. [Google Scholar] [CrossRef]
- Chun, Y.; Li, X.Y. Research progress in genetic regulation of rice panicle architecture. Chin. Bull. Bot. 2017, 52, 19–29. [Google Scholar]
- Leyser, O. Regulation of shoot branching by auxin. Trends Plant Sci. 2003, 8, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Gallavotti, A. The role of auxin in shaping shoot architecture. J. Exp. Bot. 2013, 64, 2593–2608. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, W.; Hirano, H. Antagonistic action of TILLERS ABSENT1 and FLORAL ORGAN NUMBER2 regulates stem cell maintenance during axillary meristem development in rice. New Phytol. 2020, 225, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tan, L.B.; Zhu, Z.F.; Xiao, L.T.; Xie, D.X.; Sun, C.Q. PAY1 improves plant architecture and enhances grain yield in rice. Plant J. 2015, 83, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.W.; Wang, S.H.; Li, G.H.; Wang, Q.S.; Liu, Z.H.; Yu, X.; Ding, Y.F. Effect of panicle pitrogen fertilizer on concentrations of cytokinin and auxin in young panicles of japonica rice and its relation with spikelet development. Acta Agron. Sin. 2008, 34, 2184–2189. [Google Scholar] [CrossRef]
- Liu, L.J.; Zhou, S.Q.; Liu, K.; Zhang, W.Y.; Yang, J.C. Research progress on the formation of large panicles in rice and its regulation. Acta Agron. Sin. 2023, 49, 585–596. [Google Scholar]
- Li, G.L.; Xu, B.X.; Zhang, Y.P.; Xu, Y.W.; Khan, N.U.; Xie, J.Y.; Sun, X.M.; Guo, H.F.; Wu, Z.Y.; Wang, X.Q.; et al. RGN1 controls grain number and shapes panicle architecture in rice. Plant Biotechnol. J. 2022, 20, 158–167. [Google Scholar] [CrossRef]
- Deveshwar, P.; Prusty, A.; Sharma, S.; Tyagi, A.K. Phytohormone-mediated molecular mechanisms involving multiple genes and QTL govern grain number in rice. Front. Genet. 2020, 11, 586462. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.C.; Zhao, H.; Ma, B.; Chou, S.Y.; Zhang, J.S. Diverse roles of ethylene in regulating agronomic traits in rice. Front. Plant Sci. 2017, 8, 1676. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, Y.; Mi, X.F.; Shan, J.X.; Li, X.M.; Xu, J.L.; Lin, H.X. The QTL GNP1 encodes GA20ox1, which increases grain number and yield by increasing cytokinin activity in rice panicle meristems. PLoS Genet. 2016, 12, e1006386. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.L.; Liang, W.Q.; Cui, X.; Chen, M.J.; Yin, C.S.; Luo, Z.J.; Zhu, J.Y.; Lucas, W.J.; Wang, Z.Y.; Zhang, D.B.; et al. Brassinosteroids promote development of rice pollen grains and seeds by triggering expression of Carbon Starved Anther, a MYB domain protein. Plant J. 2015, 82, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, C.C.; Li, M.; Cui, Y.R.; Shi, Y.Y.; Wu, Z.C.; Hu, Z.Q.; Wang, W.S.; Xu, J.L.; Li, Z.K. The landscape of gene–CDS–haplotype diversity in rice: Properties, population organization, footprints of domestication and breeding, and implications for genetic improvement. Mol. Plant 2021, 14, 787–804. [Google Scholar] [CrossRef]
- Alexandrov, N.; Tai, S.S.; Wang, W.S.; Mansueto, L.; Palis, K.; Fuentes, R.R.; Ulat, V.J.; Chebotarov, D.; Zhang, G.Y.; Li, Z.K.; et al. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015, 43, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, X.Y.; Zhang, L.S.; Yang, Z.T.; Xin, X.Y.; Wu, S.; Sun, C.Q.; Liu, J.X.; Yang, J.S.; Luo, X.J. Identification and characterization of OsEBS, a gene involved in enhanced plant biomass and spikelet number in rice. Plant Biotechnol. J. 2013, 11, 1044–1057. [Google Scholar] [CrossRef]
- Xu, X.Y.; E, Z.G.; Zhang, D.P.; Yun, Q.B.; Zhou, Y.; Niu, B.X.; Chen, C. OsYUC11-mediated auxin biosynthesis is essential for endosperm development of rice. Plant Physiol. 2021, 185, 934–950. [Google Scholar] [CrossRef]
- Tabuchi, H.; Zhang, Y.; Hattori, S.; Omae, M.; Shimizu-Sato, S.; Oikawa, T.; Qian, Q.; Nishimura, M.; Kitano, H.; Xie, H.; et al. LAX PANICLE2 of rice encodes a novel nuclear protein and regulates the formation of axillary meristems. Plant Cell 2011, 23, 3276–3287. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.L.; Sun, S.Y.; Wang, L.L.; Wu, Z.H.; Li, C.X.; Li, X.M.; Wang, T.; Leng, L.N.; Tian, W.S.; Lu, T.G.; et al. The RLA1/SMOS1 transcription factor functions with OsBZR1 to regulate brassinosteroid signaling and rice architecture. Plant Cell 2017, 29, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, T.; Koshioka, M.; Kojima, K.; Yoshida, H.; Kawata, M. A role of OsGA20ox1, encoding an isoform of gibberellin 20-oxidase, for regulation of plant stature in rice. Plant Mol. Biol. 2004, 55, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Kyozuka, J. Two-step regulation of LAX PANICLE1 protein accumulation in axillary meristem formation in rice. Plant Cell 2009, 21, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chuan, M.L.; Wang, H.Y.; Chen, R.J.; Tao, T.Y.; Zhou, Y.; Xu, Y.; Li, P.C.; Yao, Y.L.; Xu, C.W.; et al. Genetic and molecular factors in determining grain number per panicle of rice. Front. Plant Sci. 2022, 13, 964246. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.E.; Rashotte, A.M.; Murphy, A.S.; Normanly, J.; Tague, B.W.; Peer, W.A.; Taiz, L.; Muday, G.K. Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis. Plant Physiol. 2001, 126, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.Y.; Yin, S.X. Research progress on the effect of flavonoids on growth hormone. Mol. Plant Breed. 2018, 16, 5449–5462. [Google Scholar]
- Sanz, L.; Fernández-marcos, M.; Modrego, A.; Lewis, D.R.; Muday, G.K.; Pollmann, S.; Dueñas, M.; Celestino, S.-B.; Lorenzo, O. Nitric oxide plays a role in stem cell niche homeostasis through its interaction with auxin. Plant Physiol. 2014, 166, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.X.; Kong, W.P.; Ren, K.L.; Cheng, H. Advance of research in function of plant ABC transporters. J. Plant Genet. Resour. 2023, 14, 1–10. [Google Scholar]
- Nathan, L.M.; Ute, V.; Alexander, W.; George, J.; Duncan, B.; Anthony, B.; Malcolm, J.B.; Markus, G.; Darren, M.W.; Leah, R.B. Systems approaches reveal that ABCB and PIN proteins mediate co-dependent auxin efflux. Plant Cell 2022, 34, 2309–2327. [Google Scholar] [CrossRef]
- Su, N.N.; Zhu, A.Q.; Tao, X.; Ding, Z.J.; Chang, S.H.; Ye, F.; Zhang, Y.; Zhao, C.; Chen, Q.; Wang, J.Q.; et al. Structures and mechanisms of the Arabidopsis auxin transporter PIN3. Nature 2022, 609, 616–621. [Google Scholar] [CrossRef]
- Fujita, D.; Trijatmiko, K.R.; Tagle, A.G.; Sapasap, M.V.; Koide, Y.; Sasaki, K.; Tsakirpaloglou, N.; Gannaban, R.B.; Nishimura, T.; Yanagihara, S.; et al. NAL1 allele from a rice landrace greatly increases yield in modern indica cultivars. Proc. Natl. Acad. Sci. USA 2013, 110, 20431–20436. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.R.; Wei, X.; Huang, J.; Qiao, W.H.; Zhang, W.X.; Yang, Q.W. Study on the origin and evolution of Asian cultivated rice based on gene fragment nucleotides diversity of mitochondrial genome. J. Plant Genet. Resour. 2013, 14, 18–24. [Google Scholar]
- Lindquist, S.; Craig, E.A. The Heat-Shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Craig, E.A.; Weissman, J.S.; Horwich, A.L. Heat-Shock proteins and molecular chaperones: Mediators of protein conformation and turnover in the cell. Cell 1994, 78, 365–372. [Google Scholar] [CrossRef]
- Craig, E.A.; Gambill, B.D.; Nelson, R.J. Heat shock proteins: Molecular chaperones of protein biogenesis. Microbiol. Rev. 1993, 57, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Podlaha, O.; Webb, D.M.; Zhang, J.Z. Accelerated evolution and loss of a domain of the sperm-egg-binding protein SED1 in ancestral primates. Mol. Biol. Evol. 2006, 23, 1828–1831. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Harrison, R.A.; Renjifo, C.; Wuster, W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol. Biol. Evol. 2011, 28, 2637–2649. [Google Scholar] [CrossRef] [Green Version]
- Hillwig, M.L.; Xu, M.M.; Toyomasu, T.; Tiernan, M.S.; Wei, G.; Cui, G.H.; Huang, L.Q.; Peters, R.J. Domain loss has independently occurred multiple times in plant terpene synthase evolution. Plant J. 2011, 68, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Weiner, J.; Beaussart, F.; Bornberg-Bauer, E. Domain deletions and substitutions in the modular protein evolution. FEBS J. 2006, 273, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Saghai-Maroof, M.A.; Soliman, K.M.; Jorgensen, R.A.; Allard, R.W. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl. Acad. Sci. USA 1984, 81, 8014–8018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirén, J.; Välimäki, N.; Mäkinen, V. Indexing graphs for path queries with applications in genome research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.S.; Mauleon, R.; Hu, Z.Q.; Chebotarov, D.; Tai, S.S.; Wu, Z.C.; Li, M.; Zheng, T.Q.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | B1021 | B1022 | B1023 | G21 | G22 | G23 |
|---|---|---|---|---|---|---|
| Raw Read Number | 41,793,424 | 46,244,714 | 48,182,186 | 40,363,696 | 45,647,082 | 39,652,964 |
| Clean Read Number | 40,652,450 | 45,029,452 | 46,814,038 | 39,480,362 | 44,404,198 | 38,709,036 |
| Clean Read Rate (%) | 97.27 | 97.37 | 97.16 | 97.81 | 97.28 | 97.62 |
| Clean Base Number | 6,097,867,500 | 6,754,417,800 | 7,022,105,700 | 5,922,054,300 | 6,660,629,700 | 5,806,355,400 |
| Low-quality Read Number | 193,670 | 222,282 | 206,996 | 198,868 | 456,696 | 303,188 |
| Low-quality Read Rate (%) | 0.46 | 0.48 | 0.43 | 0.49 | 1.00 | 0.77 |
| Adapter-Polluted Read Number | 946,522 | 992,144 | 1,160,242 | 683,722 | 785,344 | 640,076 |
| Adapter-Polluted Read Rate (%) | 2.27 | 2.15 | 2.41 | 1.69 | 1.72 | 1.61 |
| Raw Q30 Base Rate (%) | 94.34 | 94.12 | 94.37 | 93.85 | 94.05 | 94.21 |
| Clean Q30 Base Rate (%) | 94.54 | 94.36 | 94.56 | 94.08 | 94.49 | 94.55 |
| GO ID | GO Term | DEGs | q Value |
|---|---|---|---|
| GO:0009734 | auxin-activated signaling pathway | 40 | 0.0335 |
| GO:0009926 | auxin polar transport | 16 | 0.0158 |
| GO:0060918 | auxin transport | 20 | 0.0014 |
| GO:0010540 | basipetal auxin transport | 11 | 0.0022 |
| GO:0003333 | amino acid transmembrane transport | 13 | 0.0014 |
| GO:0055114 | oxidation–reduction process | 79 | 0.0004 |
| GO:0009813 | flavonoid biosynthetic process | 47 | 0.0000 |
| GO:0015706 | nitrate transport | 6 | 0.0465 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, F.; Liu, M.; Dong, S.; Dong, X.; Wang, Y.; Cheng, C.; Chu, H.; Hu, Z.; Ma, F.; Yan, P.; et al. RNA-Seq Transcriptome Analysis and Evolution of OsEBS, a Gene Involved in Enhanced Spikelet Number per Panicle in Rice. Int. J. Mol. Sci. 2023, 24, 10303. https://doi.org/10.3390/ijms241210303
Niu F, Liu M, Dong S, Dong X, Wang Y, Cheng C, Chu H, Hu Z, Ma F, Yan P, et al. RNA-Seq Transcriptome Analysis and Evolution of OsEBS, a Gene Involved in Enhanced Spikelet Number per Panicle in Rice. International Journal of Molecular Sciences. 2023; 24(12):10303. https://doi.org/10.3390/ijms241210303
Chicago/Turabian StyleNiu, Fuan, Mingyu Liu, Shiqing Dong, Xianxin Dong, Ying Wang, Can Cheng, Huangwei Chu, Zejun Hu, Fuying Ma, Peiwen Yan, and et al. 2023. "RNA-Seq Transcriptome Analysis and Evolution of OsEBS, a Gene Involved in Enhanced Spikelet Number per Panicle in Rice" International Journal of Molecular Sciences 24, no. 12: 10303. https://doi.org/10.3390/ijms241210303
APA StyleNiu, F., Liu, M., Dong, S., Dong, X., Wang, Y., Cheng, C., Chu, H., Hu, Z., Ma, F., Yan, P., Lan, D., Zhang, J., Zhou, J., Sun, B., Zhang, A., Hu, J., Zhang, X., He, S., Cui, J., ... Luo, X. (2023). RNA-Seq Transcriptome Analysis and Evolution of OsEBS, a Gene Involved in Enhanced Spikelet Number per Panicle in Rice. International Journal of Molecular Sciences, 24(12), 10303. https://doi.org/10.3390/ijms241210303