

Inflammatory Bowel Disease: Crosstalk between Histamine, Immunity, and Disease


Abstract
1. Introduction
2. General Information on Histamine
2.1. Structure, Biosynthesis, and Degradation of Histamine
2.2. Function of Histamine in the Body
2.3. Sources of Histamine
2.4. Histamine Receptors

{kind=link}
| Receptor Subtype | G Protein Coupling | Expression | Molecular Mass (kDa) | Signal Transduction | References |
|---|---|---|---|---|---|
| H1 | Gαq | Smooth muscle cells of the respiratory and cardiovascular systems, endothelial cells, enterocytes, monocytes, neutrophils, T- and B-lymphocytes | 56 | PLC and PKC activation, cytosolic Ca2+ increase, protein phosphorylation and transcription of nuclear factor κB (NF-κB), nuclear factor of activated T-cells (NFAT), cyclic adenosine monophosphate (cAMP), response element binding protein (CREB), and activator protein 1 (AP-1) | [30,39] |
| H2 | Gαs | Heart tissue, enterocytes, brain cells, smooth muscle cells, T- and B-cells, and DCs | 40 | Adenylyl cyclase (AC) activation, increases cAMP, and activates protein kinase A (PKA) | [40] |
| H3 | Gi/o | Histaminergic neurons, monocytes, eosinophils | 48 | Inhibits cAMP synthesis, causes Ca2+ accumulation, and activates the mitogen-activated protein kinase (MAPK) pathway | [41,42] |
| H4 | Gi/o | Neutrophils, eosinophils, T cells, bone marrow cells, peripheral hematopoietic cells, thymus, lungs, small and large intestines, and heart | 44 | Inhibits cAMP synthesis, causes Ca2+ accumulation, and activates the mitogen-activated protein kinase (MAPK) pathway | [13] |
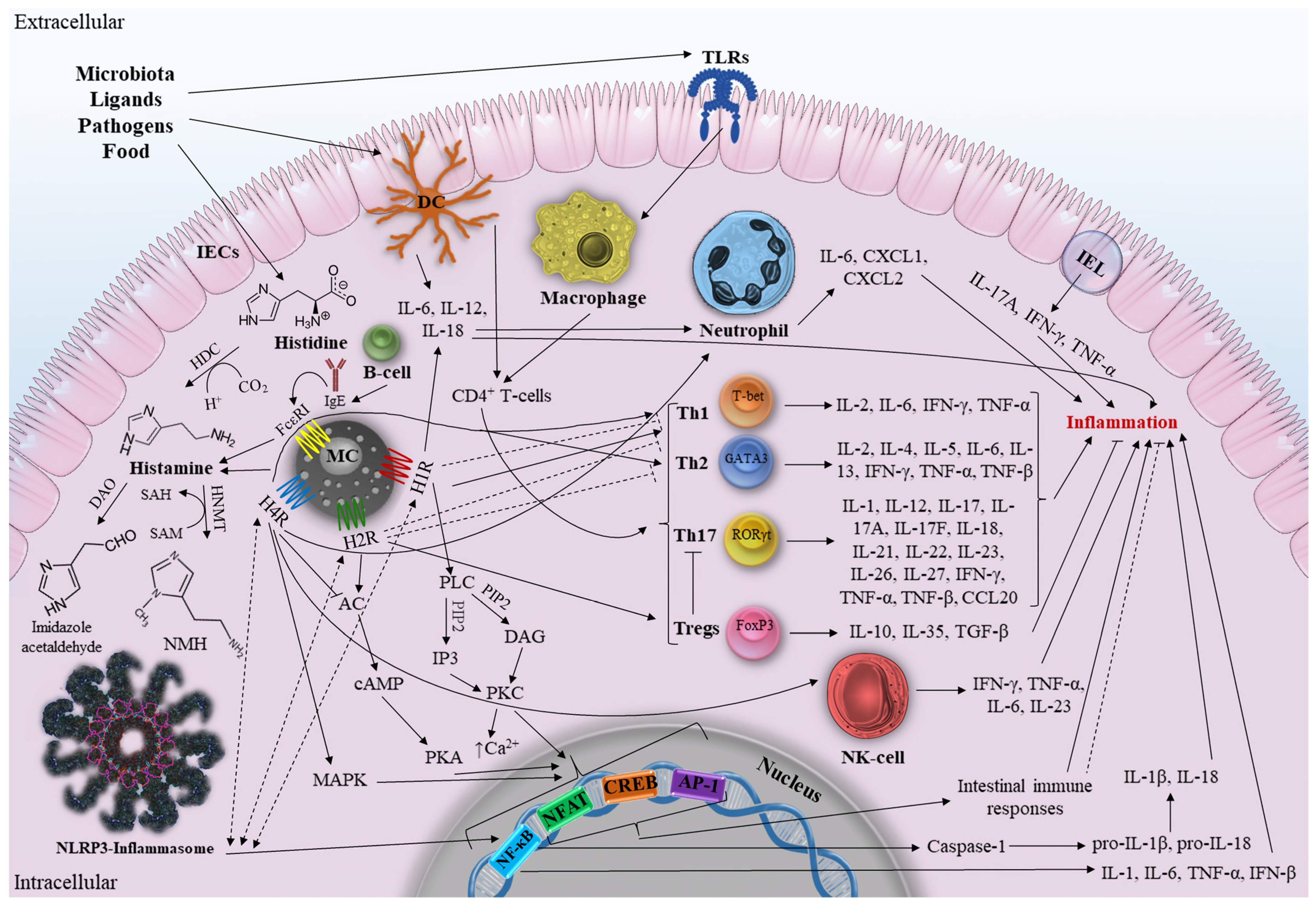
3. The Role of Histamine in Intestine
3.1. Histamine and Intestine Inflammation
3.2. General Description of Inflammatory Bowel Disease
3.3. The Role of Histamine in IBD
3.3.1. Mast Cells and IBD
Mast Cells and Adaptive Immunity
Mast Cells and Innate Immunity
Mast Cells, Enzymes, and Metabolites
3.3.2. HRs and IBD
Histamine Receptor 1
Histamine Receptor 2
Histamine Receptor 4
3.3.3. HRs Signaling Pathways in Context of IBD
4. Antagonists and Agonists of HRs—Possible IBD Therapy?
5. Polymorphism of DAO and HNMT and Impact on IBD
6. Future Perspectives and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IBD | Inflammatory bowel disease |
| CD | Crohn’s disease |
| UC | Ulcerative colitis |
| GI | Gastrointestinal tract |
| DAO | Diamine oxidase |
| HDC | L-histidine decarboxylase |
| FcεRI | FcepsilonRI |
| SP | Substance P |
| IgE | Immunoglobulin E |
| PLC | Phospholipase C |
| PIP2 | Phosphatidylinositol 4,5-diphosphate |
| DAG | Diacylglycerol |
| IP3 | Inositol-1,4,5-triphosphate |
| PKC | Protein kinase C |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| SCF | Stem cell factor |
| HNMT | Histamine-N-methyltransferase |
| SAM | S-adenosyl-L-methionine |
| NMH | N-methylhistamine |
| HIT | Histamine intolerance |
| HRs | Histamine receptors |
| GPCR | Protein-coupled receptor |
| moDCs | Monocyte-derived dendritic cells |
| MAPK | Mitogen-activated protein kinase pathway |
| FoxP3 | Tregs forkhead box P3 |
| VH | Visceral hypersensitivity |
| Ox | Oxazolone |
| AOM | Azoxymethane |
| 5-ASK | 5-Aminosalicylic Acid |
| IFX | Infliximab |
| MPO | Myeloperoxidase |
| NLR | NOD-like receptor |
| SNP | Single nucleotide polymorphism |
| IECs | Intestinal epithelial cells |
| IELs | Intestinal intraepithelial lymphocytes |
| DCs | Dendritic cells |
| Th | T-helper cells |
| Tregs | Regulatory T-cells |
| NK-cells | Natural killer T-cells |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| IFN | Interferon |
| TLRs | Toll-like receptors |
| NFAT | Nuclear factor of activated T-cells |
| cAMP | Cyclic adenosine monophosphate |
| CREB | Response element binding protein |
| AP-1 | Activator protein 1 |
| DSS | Dextran sulfate sodium |
| PAMPs | Pathogen-associated molecular patterns |
| NF-κB | Nuclear factor κB |
| PKB | Protein kinase B |
| PAR-2 | Protease-activated receptor-2 |
| mTOR | Mammalian target of rapamycin |
| APCs | Antigen-presenting cells |
| TNBS | 2,4,6-trinitrobenzenesulfonic acid |
References
- Agrawal, M.; Allin, K.H.; Petralia, F.; Colombel, J.-F.; Jess, T. Multiomics to Elucidate Inflammatory Bowel Disease Risk Factors and Pathways. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative Colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s Disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.; Zarif, R.; Mahfouz, R. Inflammatory Bowel Disease: Between Genetics and Microbiota. Mol. Biol. Rep. 2020, 47, 3053–3063. [Google Scholar] [CrossRef]
- Sultan, S.; El-Mowafy, M.; Elgaml, A.; Ahmed, T.A.E.; Hassan, H.; Mottawea, W. Metabolic Influences of Gut Microbiota Dysbiosis on Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 715506. [Google Scholar] [CrossRef]
- Kofla-Dłubacz, A.; Pytrus, T.; Akutko, K.; Sputa-Grzegrzółka, P.; Piotrowska, A.; Dzięgiel, P. Etiology of IBD—Is It Still a Mystery? Int. J. Mol. Sci. 2022, 23, 12445. [Google Scholar] [CrossRef]
- Barger, G.; Dale, H.H. Chemical Structure and Sympathomimetic Action of Amines. J. Physiol. 1910, 41, 19–59. [Google Scholar] [CrossRef]
- Hirasawa, N. Expression of Histidine Decarboxylase and Its Roles in Inflammation. Int. J. Mol. Sci. 2019, 20, 376. [Google Scholar] [CrossRef]
- Barcik, W.; Wawrzyniak, M.; Akdis, C.A.; O’Mahony, L. Immune Regulation by Histamine and Histamine-Secreting Bacteria. Curr. Opin. Immunol. 2017, 48, 108–113. [Google Scholar] [CrossRef]
- Thomas, C.M.; Hong, T.; van Pijkeren, J.P.; Hemarajata, P.; Trinh, D.V.; Hu, W.; Britton, R.A.; Kalkum, M.; Versalovic, J. Histamine Derived from Probiotic Lactobacillus Reuteri Suppresses TNF via Modulation of PKA and ERK Signaling. PLoS ONE 2012, 7, e31951. [Google Scholar] [CrossRef]
- Smolinska, S.; Groeger, D.; Perez, N.R.; Schiavi, E.; Ferstl, R.; Frei, R.; Konieczna, P.; Akdis, C.A.; Jutel, M.; O’Mahony, L. Histamine Receptor 2 Is Required to Suppress Innate Immune Responses to Bacterial Ligands in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 1575–1586. [Google Scholar] [CrossRef]
- Schirmer, B.; Neumann, D. The Function of the Histamine H4 Receptor in Inflammatory and Inflammation-Associated Diseases of the Gut. Int. J. Mol. Sci. 2021, 22, 6116. [Google Scholar] [CrossRef]
- Sander, L.E.; Lorentz, A.; Sellge, G.; Coëffier, M.; Neipp, M.; Veres, T.; Frieling, T.; Meier, P.N.; Manns, M.P.; Bischoff, S.C. Selective Expression of Histamine Receptors H1R, H2R, and H4R, but Not H3R, in the Human Intestinal Tract. Gut 2006, 55, 498–504. [Google Scholar] [CrossRef]
- Jutel, M.; Blaser, K.; Akdis, C.A. The Role of Histamine in Regulation of Immune Responses. Chem. Immunol. Allergy 2006, 91, 174–187. [Google Scholar] [CrossRef]
- Beermann, S.; Bernhardt, G.; Seifert, R.; Buschauer, A.; Neumann, D. Histamine H(1)- and H(4)-Receptor Signaling Cooperatively Regulate MAPK Activation. Biochem. Pharm. 2015, 98, 432–439. [Google Scholar] [CrossRef]
- Shah, R.; Richardson, P.; Yu, H.; Kramer, J.; Hou, J.K. Gastric Acid Suppression Is Associated with an Increased Risk of Adverse Outcomes in Inflammatory Bowel Disease. Digestion 2017, 95, 188–193. [Google Scholar] [CrossRef]
- D’sa, F.F.; Fernandes, E.Z.; Kesarkar, S.V.; Swaminathan, L.; Kunhikatta, V.; Rashid, M.; Thunga, G.; Chandran, V.P.; Nair, S. Use of Histamine-2 Receptor Antagonists and Risk of Inflammatory Bowel Diseases: A Systematic Review and Meta-Analysis of Observational Studies. J. Clin. Pharm. Ther. 2022, 47, 1103–1111. [Google Scholar] [CrossRef]
- Ferstl, R.; Akdis, C.A.; O’Mahony, L. Histamine Regulation of Innate and Adaptive Immunity. Front. Biosci. 2012, 17, 40–53. [Google Scholar] [CrossRef]
- Yan, J.-B.; Luo, M.-M.; Chen, Z.-Y.; He, B.-H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 8813558. [Google Scholar] [CrossRef]
- Damaj, B.B.; Becerra, C.B.; Esber, H.J.; Wen, Y.; Maghazachi, A.A. Functional Expression of H4 Histamine Receptor in Human Natural Killer Cells, Monocytes, and Dendritic Cells. J. Immunol. 2007, 179, 7907–7915. [Google Scholar] [CrossRef] [PubMed]
- Tiligada, E.; Ennis, M. Histamine Pharmacology: From Sir Henry Dale to the 21st Century. Br. J. Pharm. 2020, 177, 469–489. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, Y.; Liang, J.; Finkelman, F.D. Molecular Regulation of Histamine Synthesis. Front. Immunol. 2018, 9, 1392. [Google Scholar] [CrossRef] [PubMed]
- Mirzahosseini, A.; Dalmadi, B.; Csutora, P. Histamine Receptor H4 Regulates Mast Cell Degranulation and IgE Induced FcεRI Upregulation in Murine Bone Marrow-Derived Mast Cells. Cell. Immunol. 2013, 283, 38–44. [Google Scholar] [CrossRef]
- Elmore, B.O.; Bollinger, J.A.; Dooley, D.M. Human Kidney Diamine Oxidase: Heterologous Expression, Purification, and Characterization. J. Biol. Inorg. Chem. 2002, 7, 565–579. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, H.-H.; Liu, Z.-Q.; Chen, X.-P.; He, Y.-J. Chapter 44—Pharmacogenomics in China. In Handbook of Pharmacogenomics and Stratified Medicine; Padmanabhan, S., Ed.; Academic Press: San Diego, CA, USA, 2014; pp. 999–1013. ISBN 978-0-12-386882-4. [Google Scholar]
- Schnedl, W.J.; Enko, D. Histamine Intolerance Originates in the Gut. Nutrients 2021, 13, 1262. [Google Scholar] [CrossRef]
- Sgambellone, S.; Marri, S.; Masini, E.; Lucarini, L. New Insight in Histamine Functions. Biomolecules 2022, 12, 609. [Google Scholar] [CrossRef]
- Branco, A.C.C.C.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of Histamine in Modulating the Immune Response and Inflammation. Mediat. Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef]
- Smolinska, S.; Winiarska, E.; Globinska, A.; Jutel, M. Histamine: A Mediator of Intestinal Disorders—A Review. Metabolites 2022, 12, 895. [Google Scholar] [CrossRef]
- Lieberman, P. The Basics of Histamine Biology. Ann. Allergy Asthma Immunol. 2011, 106, S2–S5. [Google Scholar] [CrossRef]
- Borriello, F.; Iannone, R.; Marone, G. Histamine Release from Mast Cells and Basophils. In Histamine and Histamine Receptors in Health and Disease; Hattori, Y., Seifert, R., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2017; pp. 121–139. ISBN 978-3-319-58194-1. [Google Scholar]
- Gagic, M.; Jamroz, E.; Krizkova, S.; Milosavljevic, V.; Kopel, P.; Adam, V. Current Trends in Detection of Histamine in Food and Beverages. J. Agric. Food Chem. 2019, 67, 773–783. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Jin, H.; Chen, L.; Ji, J.; Zhang, Z. Histamine Intolerance—A Kind of Pseudoallergic Reaction. Biomolecules 2022, 12, 454. [Google Scholar] [CrossRef]
- Smolinska, S.; Jutel, M.; Crameri, R.; O’Mahony, L. Histamine and Gut Mucosal Immune Regulation. Allergy 2014, 69, 273–281. [Google Scholar] [CrossRef]
- Panula, P. Chapter 23—Histamine Receptors, Agonists, and Antagonists in Health and Disease. In Handbook of Clinical Neurology; Swaab, D.F., Kreier, F., Lucassen, P.J., Salehi, A., Buijs, R.M., Eds.; The Human Hypothalamus; Elsevier: Amsterdam, The Netherlands, 2021; Volume 180, pp. 377–387. [Google Scholar] [CrossRef]
- Falkenstein, M.; Elek, M.; Stark, H. Chemical Probes for Histamine Receptor Subtypes. In The Functional Roles of Histamine Receptors; Yanai, K., Passani, M.B., Eds.; Current Topics in Behavioral Neurosciences; Springer International Publishing: Cham, Switzerland, 2022; pp. 29–76. ISBN 978-3-031-16997-7. [Google Scholar]
- Mizuguchi, H.; Kitamura, Y.; Takeda, N.; Fukui, H. Molecular Signaling and Transcriptional Regulation of Histamine H1 Receptor Gene. In The Functional Roles of Histamine Receptors; Yanai, K., Passani, M.B., Eds.; Current Topics in Behavioral Neurosciences; Springer International Publishing: Cham, Switzerland, 2022; pp. 91–110. ISBN 978-3-031-16997-7. [Google Scholar]
- Díaz Nebreda, A.; Zappia, C.D.; Rodríguez González, A.; Sahores, A.; Sosa, M.; Burghi, V.; Monczor, F.; Davio, C.; Fernández, N.; Shayo, C. Involvement of Histamine H1 and H2 Receptor Inverse Agonists in Receptor’s Crossregulation. Eur. J. Pharmacol. 2019, 847, 42–52. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Role of the Histamine H3 Receptor in the Central Nervous System. Handb. Exp. Pharm. 2017, 241, 277–299. [Google Scholar] [CrossRef]
- Wulff, B.S.; Hastrup, S.; Rimvall, K. Characteristics of Recombinantly Expressed Rat and Human Histamine H3 Receptors. Eur. J. Pharm. 2002, 453, 33–41. [Google Scholar] [CrossRef]
- Church, M.K. Allergy, Histamine and Antihistamines. Handb. Exp. Pharm. 2017, 241, 321–331. [Google Scholar] [CrossRef]
- Deiteren, A.; Man, J.G.D.; Ruyssers, N.E.; Moreels, T.G.; Pelckmans, P.A.; Winter, B.Y.D. Histamine H4 and H1 Receptors Contribute to Postinflammatory Visceral Hypersensitivity. Gut 2014, 63, 1873–1882. [Google Scholar] [CrossRef]
- Mehandru, S.; Colombel, J.-F. The Intestinal Barrier, an Arbitrator Turned Provocateur in IBD. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 83–84. [Google Scholar] [CrossRef]
- Regner, E.H.; Ohri, N.; Stahly, A.; Gerich, M.E.; Fennimore, B.P.; Ir, D.; Jubair, W.K.; Görg, C.; Siebert, J.; Robertson, C.E.; et al. Functional Intraepithelial Lymphocyte Changes in Inflammatory Bowel Disease and Spondyloarthritis Have Disease Specific Correlations with Intestinal Microbiota. Arthritis Res. Ther. 2018, 20, 149. [Google Scholar] [CrossRef] [PubMed]
- McKernan, D.P. Toll-Like Receptors as Drug Targets in the Intestinal Epithelium. Handb. Exp. Pharm. 2022, 276, 291–314. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, K.G.; Desai, D.C.; Ashavaid, T.F.; Dherai, A.J. Microbiome and Metabolome in Inflammatory Bowel Disease. J. Gastroenterol. Hepatol. 2023, 38, 34–43. [Google Scholar] [CrossRef]
- Boeckxstaens, G. Mast Cells and Inflammatory Bowel Disease. Curr. Opin. Pharmacol. 2015, 25, 45–49. [Google Scholar] [CrossRef]
- Fox, C.C.; Lichtenstein, L.M.; Roche, J.K. Intestinal Mast Cell Responses in Idiopathic Inflammatory Bowel Disease. Histamine Release from Human Intestinal Mast Cells in Response to Gut Epithelial Proteins. Dig. Dis. Sci. 1993, 38, 1105–1112. [Google Scholar] [CrossRef]
- Gelbmann, C.M.; Mestermann, S.; Gross, V.; Köllinger, M.; Schölmerich, J.; Falk, W. Strictures in Crohn’s Disease Are Characterised by an Accumulation of Mast Cells Colocalised with Laminin but Not with Fibronectin or Vitronectin. Gut 1999, 45, 210–217. [Google Scholar] [CrossRef]
- He, S.-H. Key Role of Mast Cells and Their Major Secretory Products in Inflammatory Bowel Disease. World J. Gastroenterol. 2004, 10, 309–318. [Google Scholar] [CrossRef]
- Araki, Y.; Andoh, A.; Fujiyama, Y.; Bamba, T. Development of Dextran Sulphate Sodium-Induced Experimental Colitis Is Suppressed in Genetically Mast Cell-Deficient Ws/Ws Rats. Clin. Exp. Immunol. 2000, 119, 264–269. [Google Scholar] [CrossRef]
- Zhao, P.; Dong, L.; Luo, J.; Guan, H.; Ma, H.; Wang, X. Possible Role of Mast Cells and Neuropeptides in the Recovery Process of Dextran Sulfate Sodium-Induced Colitis in Rats. Chin. Med. Sci. J. 2013, 28, 28–33. [Google Scholar] [CrossRef]
- Zhang, Z.; Kurashima, Y. Two Sides of the Coin: Mast Cells as a Key Regulator of Allergy and Acute/Chronic Inflammation. Cells 2021, 10, 1615. [Google Scholar] [CrossRef]
- Bene, L.; Sápi, Z.; Bajtai, A.; Buzás, E.; Szentmihályi, A.; Arató, A.; Tulassay, Z.; Falus, A. Partial Protection against Dextran Sodium Sulphate Induced Colitis in Histamine-Deficient, Histidine Decarboxylase Knockout Mice. J. Pediatr. Gastroenterol. Nutr. 2004, 39, 171–176. [Google Scholar] [CrossRef]
- Wu, L.; Feng, B.-S.; He, S.-H.; Zheng, P.-Y.; Croitoru, K.; Yang, P.-C. Bacterial Peptidoglycan Breaks down Intestinal Tolerance via Mast Cell Activation: The Role of TLR2 and NOD2. Immunol. Cell Biol. 2007, 85, 538–545. [Google Scholar] [CrossRef]
- Okumura, S.; Yuki, K.; Kobayashi, R.; Okamura, S.; Ohmori, K.; Saito, H.; Ra, C.; Okayama, Y. Hyperexpression of NOD2 in Intestinal Mast Cells of Crohn’s Disease Patients: Preferential Expression of Inflammatory Cell-Recruiting Molecules via NOD2 in Mast Cells. Clin. Immunol. 2009, 130, 175–185. [Google Scholar] [CrossRef]
- Liu, B.; Yang, M.-Q.; Yu, T.-Y.; Yin, Y.-Y.; Liu, Y.; Wang, X.-D.; He, Z.-G.; Yin, L.; Chen, C.-Q.; Li, J.-Y. Mast Cell Tryptase Promotes Inflammatory Bowel Disease–Induced Intestinal Fibrosis. Inflamm. Bowel Dis. 2021, 27, 242–255. [Google Scholar] [CrossRef]
- Winterkamp, S.; Weidenhiller, M.; Otte, P.; Stolper, J.; Schwab, D.; Hahn, E.G.; Raithel, M. Urinary Excretion of N-Methylhistamine as a Marker of Disease Activity in Inflammatory Bowel Disease. Am. J. Gastroenterol. 2002, 97, 3071–3077. [Google Scholar] [CrossRef]
- Anfinsen, K.P.; Berghoff, N.; Priestnall, S.L.; Suchodolski, J.S.; Steiner, J.M.; Allenspach, K. Urinary and Faecal N-Methylhistamine Concentrations Do Not Serve as Markers for Mast Cell Activation or Clinical Disease Activity in Dogs with Chronic Enteropathies. Acta Vet. Scand. 2014, 56, 90. [Google Scholar] [CrossRef]
- Breunig, E.; Michel, K.; Zeller, F.; Seidl, S.; Weyhern, C.W.H.v.; Schemann, M. Histamine Excites Neurones in the Human Submucous Plexus through Activation of H1, H2, H3 and H4 Receptors. J. Physiol. 2007, 583, 731–742. [Google Scholar] [CrossRef]
- Homaidan, F.R.; Tripodi, J.; Zhao, L.; Burakoff, R. Regulation of Ion Transport by Histamine in Mouse Cecum. Eur. J. Pharmacol. 1997, 331, 199–204. [Google Scholar] [CrossRef]
- Frei, R.; Ferstl, R.; Konieczna, P.; Ziegler, M.; Simon, T.; Rugeles, T.M.; Mailand, S.; Watanabe, T.; Lauener, R.; Akdis, C.A.; et al. Histamine Receptor 2 Modifies Dendritic Cell Responses to Microbial Ligands. J. Allergy Clin. Immunol. 2013, 132, 194–204. [Google Scholar] [CrossRef]
- Gao, C.; Major, A.; Rendon, D.; Lugo, M.; Jackson, V.; Shi, Z.; Mori-Akiyama, Y.; Versalovic, J. Histamine H2 Receptor-Mediated Suppression of Intestinal Inflammation by Probiotic Lactobacillus Reuteri. mBio 2015, 6, e01358-15. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, B.; Rezniczek, T.; Seifert, R.; Neumann, D. Proinflammatory Role of the Histamine H4 Receptor in Dextrane Sodium Sulfate-Induced Acute Colitis. Biochem. Pharmacol. 2015, 98, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Varga, C.; Horvath, K.; Berko, A.; Thurmond, R.L.; Dunford, P.J.; Whittle, B.J.R. Inhibitory Effects of Histamine H4 Receptor Antagonists on Experimental Colitis in the Rat. Eur. J. Pharmacol. 2005, 522, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, J.B.; Szabo, A.; Hsu, C.-L.; Krier-Burris, R.; Schroeder, H.; Wang, M.Y.; Carter, R.; Velez, T.; Aguiniga, L.M.; Brown, J.B.; et al. Histamine Drives Severity of Innate Inflammation via Histamine 4 Receptor in Murine Experimental Colitis. Mucosal Immunol. 2018, 11, 861–870. [Google Scholar] [CrossRef]
- Wunschel, E.J.; Schirmer, B.; Seifert, R.; Neumann, D. Lack of Histamine H4-Receptor Expression Aggravates TNBS-Induced Acute Colitis Symptoms in Mice. Front. Pharm. 2017, 8, 642. [Google Scholar] [CrossRef]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-ΚB in Inflammatory Bowel Disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef]
- Li, R.; Zhu, S. NLRP6 Inflammasome. Mol. Asp. Med. 2020, 76, 100859. [Google Scholar] [CrossRef]
- Jutel, M.; Watanabe, T.; Klunker, S.; Akdis, M.; Thomet, O.A.; Malolepszy, J.; Zak-Nejmark, T.; Koga, R.; Kobayashi, T.; Blaser, K.; et al. Histamine Regulates T-Cell and Antibody Responses by Differential Expression of H1 and H2 Receptors. Nature 2001, 413, 420–425. [Google Scholar] [CrossRef]
- Dy, M.; Schneider, E. Histamine–Cytokine Connection in Immunity and Hematopoiesis. Cytokine Growth Factor Rev. 2004, 15, 393–410. [Google Scholar] [CrossRef]
- Mi, J.; Liu, Z.; Pei, S.; Wu, X.; Zhao, N.; Jiang, L.; Zhang, Z.; Bai, X. Mendelian Randomization Study for the Roles of IL-18 and IL-1 Receptor Antagonist in the Development of Inflammatory Bowel Disease. Int. Immunopharmacol. 2022, 110, 109020. [Google Scholar] [CrossRef]
- Matsuno, H.; Kayama, H.; Nishimura, J.; Sekido, Y.; Osawa, H.; Barman, S.; Ogino, T.; Takahashi, H.; Haraguchi, N.; Hata, T.; et al. CD103+ Dendritic Cell Function Is Altered in the Colons of Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1524–1534. [Google Scholar] [CrossRef]
- Butcher, M.J.; Zhu, J. Recent Advances in Understanding the Th1/Th2 Effector Choice. Fac. Rev. 2021, 10, 30. [Google Scholar] [CrossRef]
- Cohen, N.A.; Rubin, D.T. New Targets in Inflammatory Bowel Disease Therapy: 2021. Curr. Opin. Gastroenterol. 2021, 37, 357–363. [Google Scholar] [CrossRef]
- Isaacs, M.J.; Tharp, M.D. 32—Antihistamines. In Comprehensive Dermatologic Drug Therapy, 4th ed.; Wolverton, S.E., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 349–357.e2. ISBN 978-0-323-61211-1. [Google Scholar]
- Neumann, D.; Seifert, R. The Therapeutic Potential of Histamine Receptor Ligands in Inflammatory Bowel Disease. Biochem. Pharmacol. 2014, 91, 12–17. [Google Scholar] [CrossRef]
- Raithel, M.; Nägel, A.; Zopf, Y.; deRossi, T.; Stengel, C.; Hagel, A.; Kressel, J.; Hahn, E.G.; Konturek, P. Plasma Histamine Levels (H) during Adjunctive H1-Receptor Antagonist Treatment with Loratadine in Patients with Active Inflammatory Bowel Disease (IBD). Inflamm. Res. 2010, 59 (Suppl. 2), S257–S258. [Google Scholar] [CrossRef]
- Church, M.K.; Frischbutter, S.; Kolkhir, P.; Maurer, M. 5.28—The Pharmacology of Antihistamines. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Oxford, UK, 2022; pp. 515–535. ISBN 978-0-12-820876-2. [Google Scholar]
- Fogel, W.A.; Jochem, J.; Lewinski, A. Influence of the H3/H4 Receptor Antagonist, Thioperamide on Regional Haemodynamics in Rats with Trinitrobenzene Sulfonic Acid-Induced Colitis. Inflamm. Res. 2007, 56 (Suppl. 1), S21–S22. [Google Scholar] [CrossRef]
- Zampeli, E.; Tiligada, E. The Role of Histamine H4 Receptor in Immune and Inflammatory Disorders. Br. J. Pharm. 2009, 157, 24–33. [Google Scholar] [CrossRef]
- Deiteren, A.; De Man, J.G.; Pelckmans, P.A.; De Winter, B.Y. Histamine H4 Receptors in the Gastrointestinal Tract. Br. J. Pharmacol. 2015, 172, 1165–1178. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Ohnuma, S.; Suzuki, H.; Ishida, M.; Ishii, K.; Hirosawa, T.; Hirashima, T.; Murakami, M.; Kobayashi, M.; Kudoh, K.; et al. Infliximab Inhibits Colitis Associated Cancer in Model Mice by Downregulating Genes Associated with Mast Cells and Decreasing Their Accumulation. Curr. Issues Mol. Biol. 2023, 45, 2895–2907. [Google Scholar] [CrossRef]
- Lichtenstein, L.; Ron, Y.; Kivity, S.; Ben-Horin, S.; Israeli, E.; Fraser, G.M.; Dotan, I.; Chowers, Y.; Confino-Cohen, R.; Weiss, B. Infliximab-Related Infusion Reactions: Systematic Review. J. Crohns. Colitis. 2015, 9, 806–815. [Google Scholar] [CrossRef]
- Bermejo, F.; López San Román, A.; Algaba, A.; van Domselaar, M.; Carneros, J.A.; Rivero, M.; Piqueras, B.; Valer, M.P. Efficacy of premedication with intravenous corticosteroids and antihistaminics in preventing infusion reactions to infliximab]. Gastroenterol. Hepatol. 2008, 31, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Poh, J.; Knowles, S. Safety of 5-Aminosalicylic Acid Derivatives in Patients with Sensitivity to Acetylsalicylic Acid and Nonsteroidal Anti-Inflammatory Drugs. Can. J. Hosp. Pharm. 2014, 67, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Raithel, M.; Schwelberger, H.G. Analysis of Diamine Oxidase Gene Polymorphisms in Patients with Inflammatory Bowel Disease. Inflamm. Res. 2001, 50 (Suppl. 2), S68–S69. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Raithel, M.; Schwelberger, H.G. Histamine N-Methyltransferase and Diamine Oxidase Gene Polymorphisms in Patients with Inflammatory and Neoplastic Intestinal Diseases. Inflamm. Res. 2002, 51 (Suppl. 1), S91–S92. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Raithel, M.; Schwelberger, H.G. Characterisation of Functional Polymorphisms of the Human Diamine Oxidase Gene. Inflamm. Res. 2005, 54 (Suppl. 1), S58–S59. [Google Scholar] [CrossRef]
- García-Martín, E.; Mendoza, J.L.; Martínez, C.; Taxonera, C.; Urcelay, E.; Ladero, J.M.; de la Concha, E.G.; Díaz-Rubio, M.; Agúndez, J.A. Severity of Ulcerative Colitis Is Associated with a Polymorphism at Diamine Oxidase Gene but Not at Histamine N-Methyltransferase Gene. World J. Gastroenterol. 2006, 12, 615–620. [Google Scholar] [CrossRef]
- Honzawa, Y.; Nakase, H.; Matsuura, M.; Chiba, T. Clinical Significance of Serum Diamine Oxidase Activity in Inflammatory Bowel Disease: Importance of Evaluation of Small Intestinal Permeability. Inflamm. Bowel Dis. 2011, 17, E23–E25. [Google Scholar] [CrossRef]
- López Palacios, N.; Agúndez, J.A.G.; Mendoza, J.L.; García-Martín, E.; Martínez, C.; Fuentes Ferrer, M.E.; Ladero, J.M.; Taxonera, C.; Díaz-Rubio, M. Analysis of a Non-Synonymous Single Nucleotide Polymorphism of the Human Diamine Oxidase Gene (Ref. SNP ID: Rs1049793) in Patients with Crohn’s Disease. Scand. J. Gastroenterol. 2009, 44, 1207–1212. [Google Scholar] [CrossRef]
- Li, J.; Sun, C.; Cai, W.; Li, J.; Rosen, B.P.; Chen, J. Insights into S-Adenosyl-l-Methionine (SAM)-Dependent Methyltransferase Related Diseases and Genetic Polymorphisms. Mutat. Res. Rev. Mutat. Res. 2021, 788, 108396. [Google Scholar] [CrossRef]
- Hailong, C.; Mei, Q.; Zhang, L.; Xu, J. C314T Polymorphism in Histamine N-Methyltransferase Gene and Susceptibility to Duodenal Ulcer in Chinese Population. Clin. Chim. Acta 2008, 389, 51–54. [Google Scholar] [CrossRef]

| Sources | Site of Production | References |
|---|---|---|
| Endogenous | Immune and nonimmune cells: endothelial cells, nerve cells, histaminergic neurons, intestinal epithelial cells (IECs), neutrophils, eosinophils, monocytes, macrophages, DCs, T- and B-cells, and Langerhans cells | [32,33] |
| Exogenous | Food: cheese, wine, sauerkraut, soy sauce, jerky, and seafood; Bacteria: Escherichia coli, Lactobacillus vaginalis, Lactobacillus reuteri, Morganella morganii, Hafnia alvei, Proteus vulgaris, Proteus milabilis, Enterobacter aerogenes, Raoultella planticola, Raoultella ornithinolytica, Citrobacter freundii, Pseudomonas fluorescens, and Photobacterium damselae | [10,11,34,35,36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvornikova, K.A.; Platonova, O.N.; Bystrova, E.Y. Inflammatory Bowel Disease: Crosstalk between Histamine, Immunity, and Disease. Int. J. Mol. Sci. 2023, 24, 9937. https://doi.org/10.3390/ijms24129937
Dvornikova KA, Platonova ON, Bystrova EY. Inflammatory Bowel Disease: Crosstalk between Histamine, Immunity, and Disease. International Journal of Molecular Sciences. 2023; 24(12):9937. https://doi.org/10.3390/ijms24129937
Chicago/Turabian StyleDvornikova, Kristina A., Olga N. Platonova, and Elena Y. Bystrova. 2023. "Inflammatory Bowel Disease: Crosstalk between Histamine, Immunity, and Disease" International Journal of Molecular Sciences 24, no. 12: 9937. https://doi.org/10.3390/ijms24129937
APA StyleDvornikova, K. A., Platonova, O. N., & Bystrova, E. Y. (2023). Inflammatory Bowel Disease: Crosstalk between Histamine, Immunity, and Disease. International Journal of Molecular Sciences, 24(12), 9937. https://doi.org/10.3390/ijms24129937