
Adaptive Changes in Detoxification Metabolism and Transmembrane Transport of Bombyx mori Malpighian Tubules to Artificial Diet
 , and
, and
Abstract
:1. Introduction
2. Results
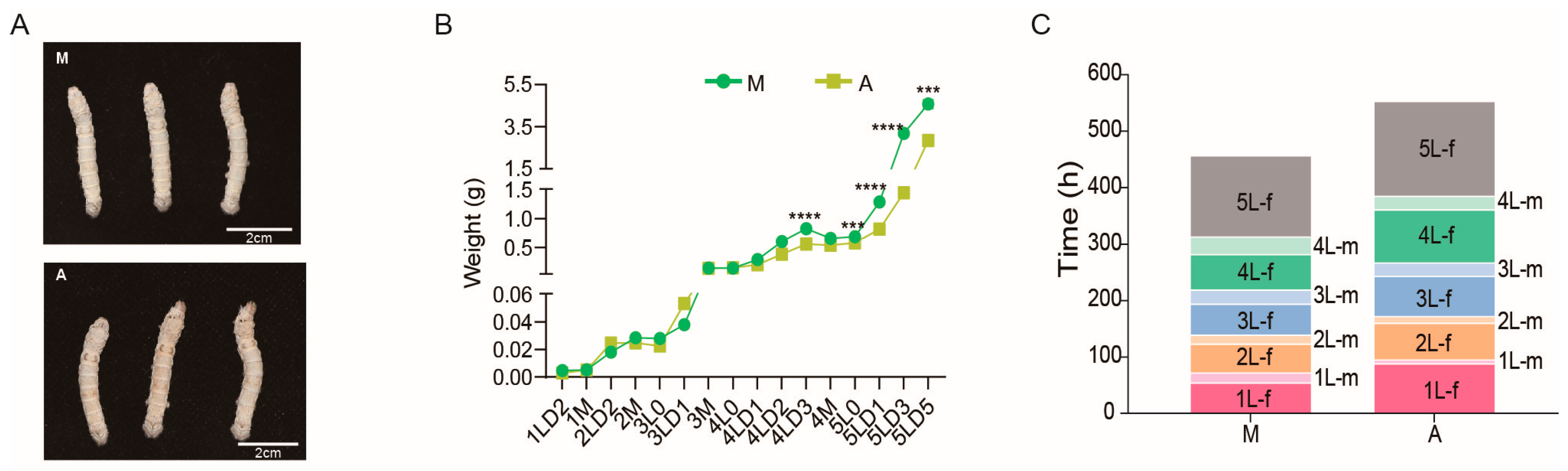
2.1. Effect of Diet on Silkworm Growth and Development Performance
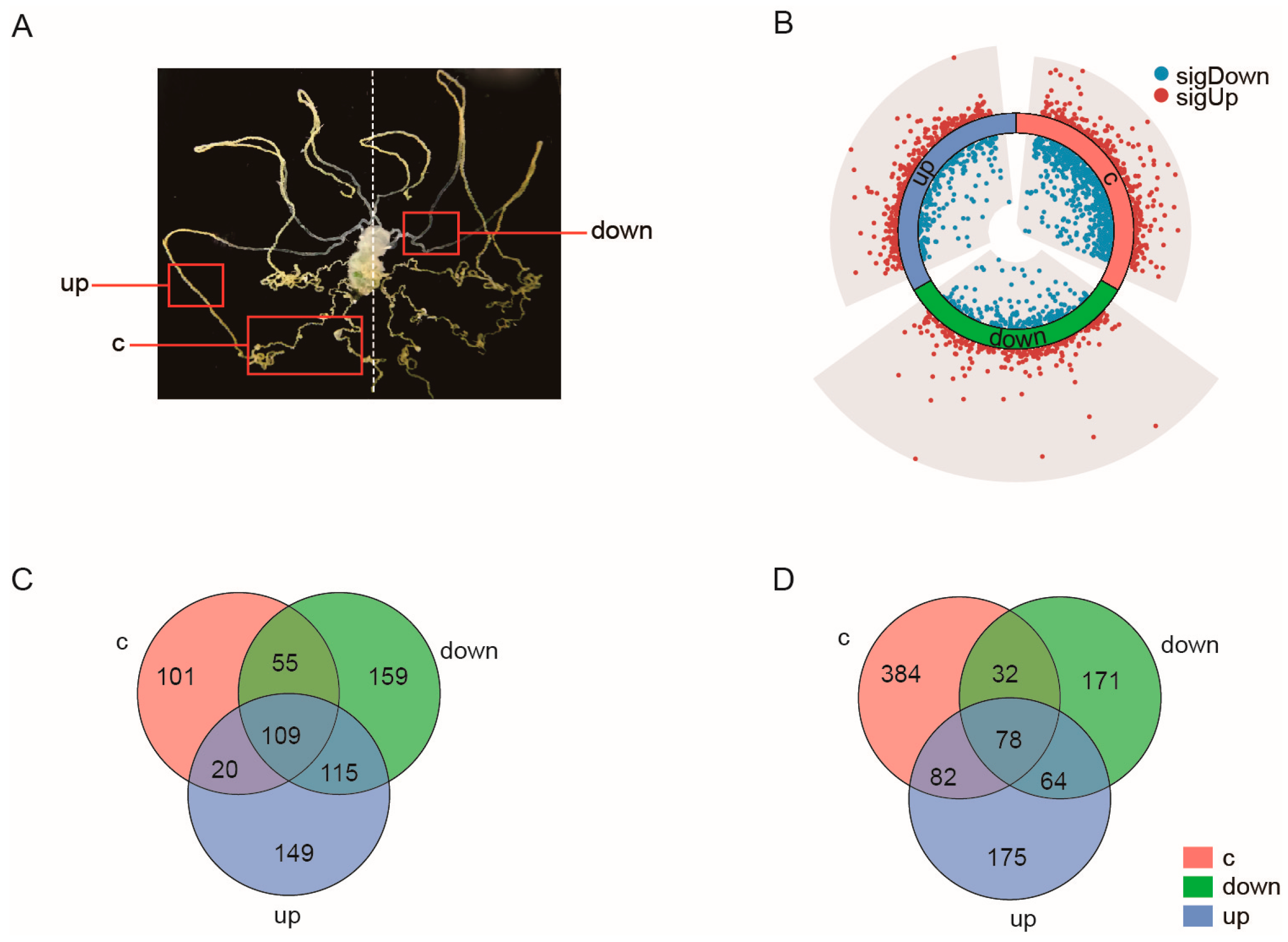
2.2. Malpighian Tubules Show Distinct Transcriptomic Alterations in Response to Diet Change
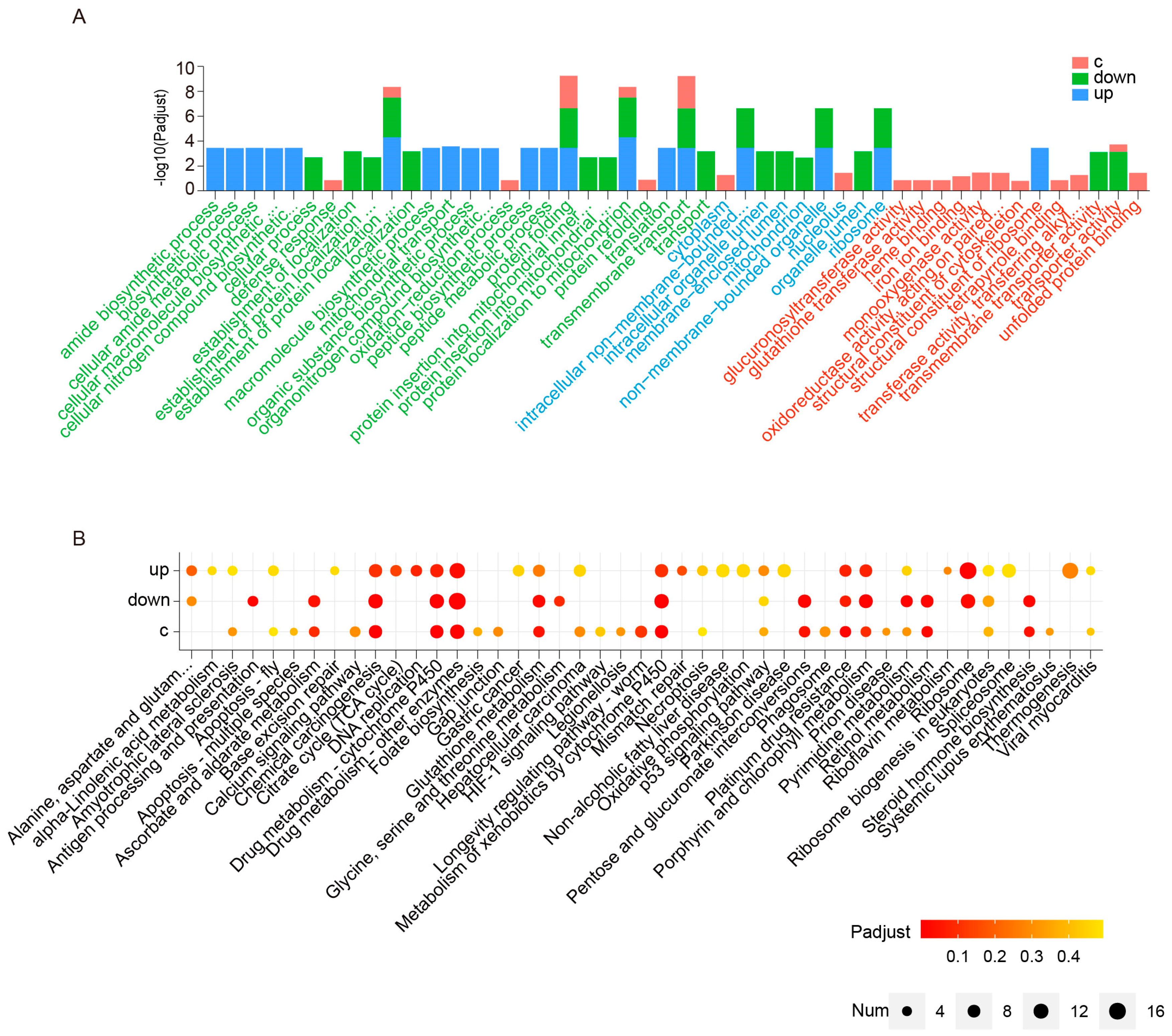
2.3. Analysis of the Main DEGs
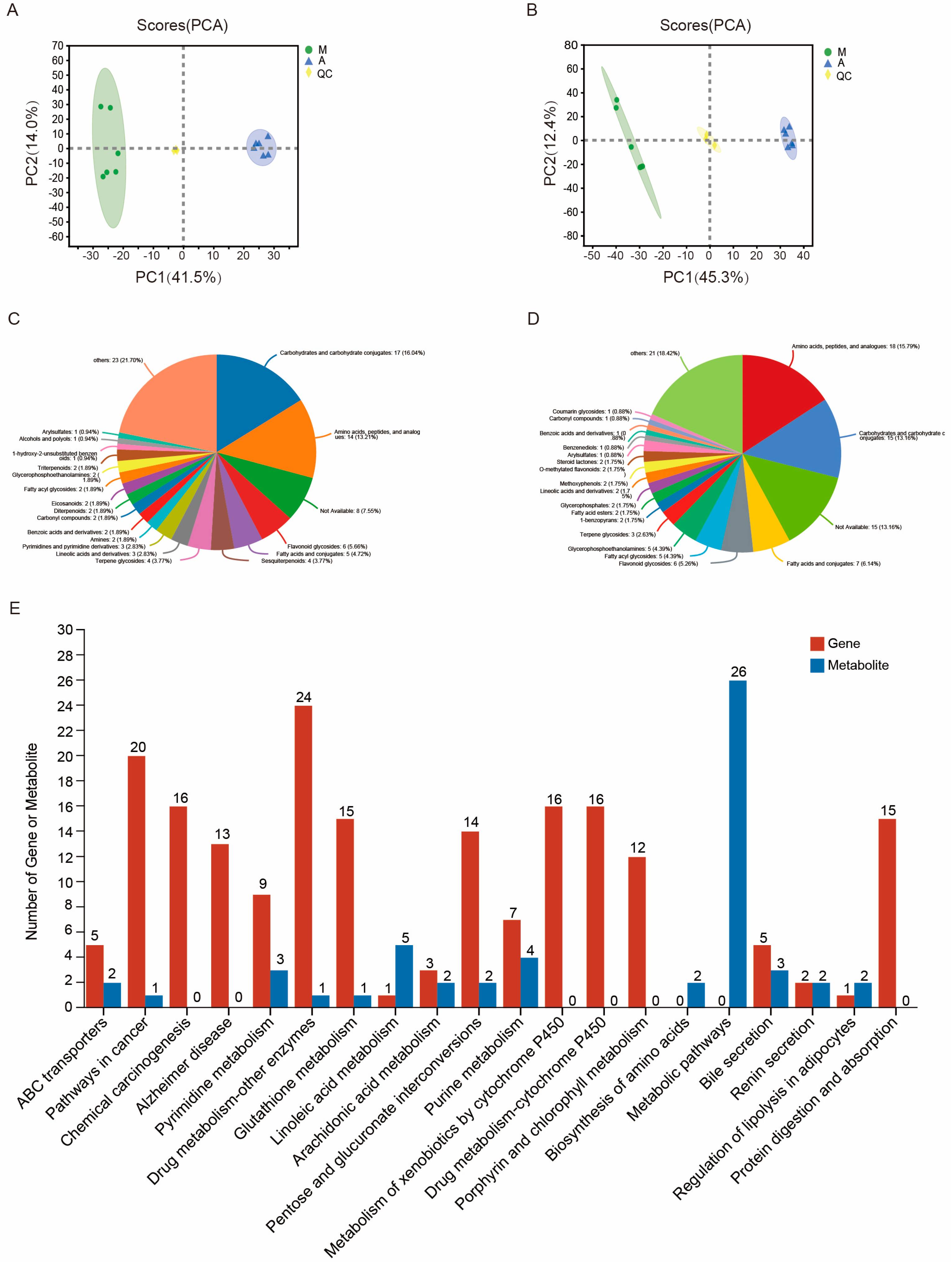
2.4. Differentially Abundant Metabolites and Comparative Analysis of Transcription and Metabolism in the Malpighian Tubules in Response to Artificial Diet Feeding
3. Discussion
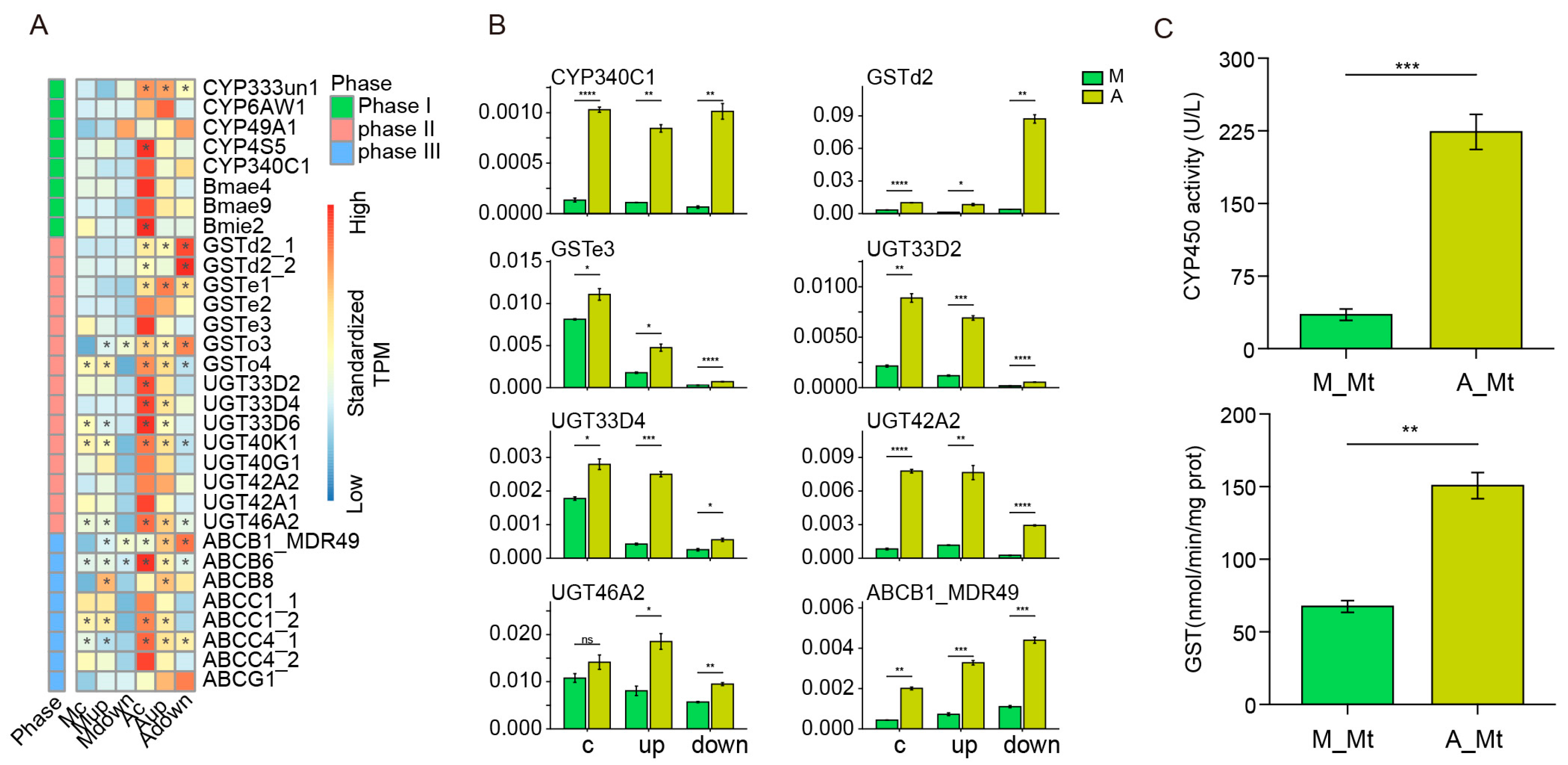
3.1. Activation of a Fully Mobilized Detoxifying Enzyme System for Metabolic Adaptation to an Artificial Diet
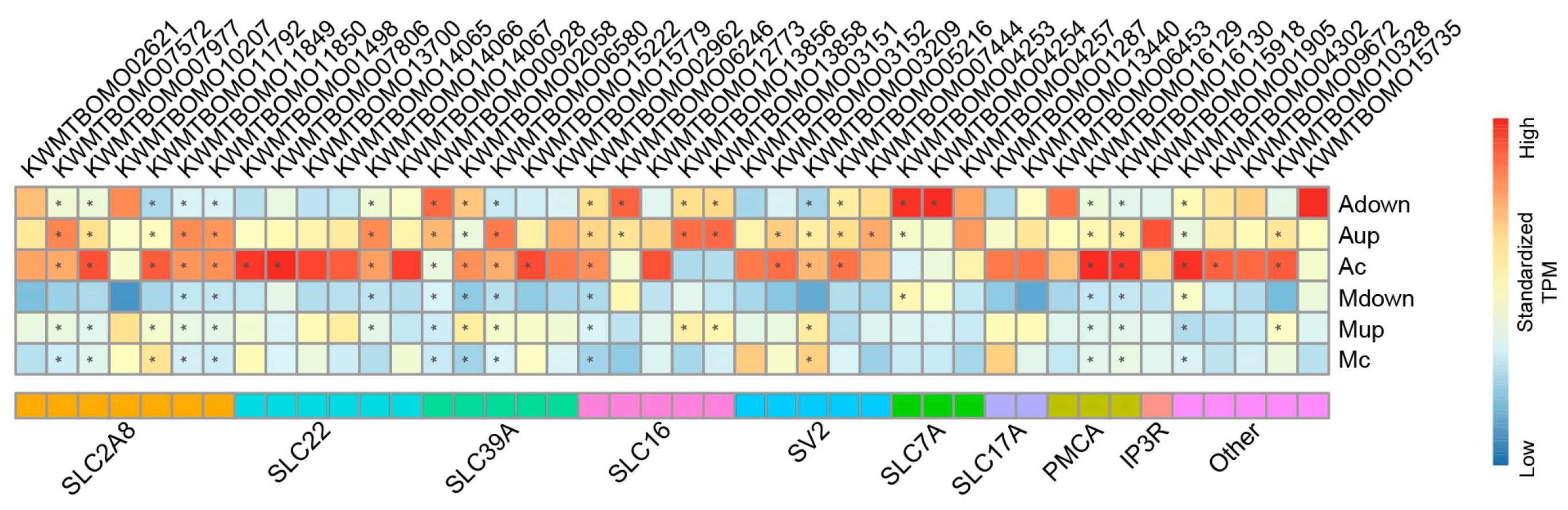
3.2. Artificial Diets Induce Transmembrane Transport Function in the Malpighian Tubules of Silkworms
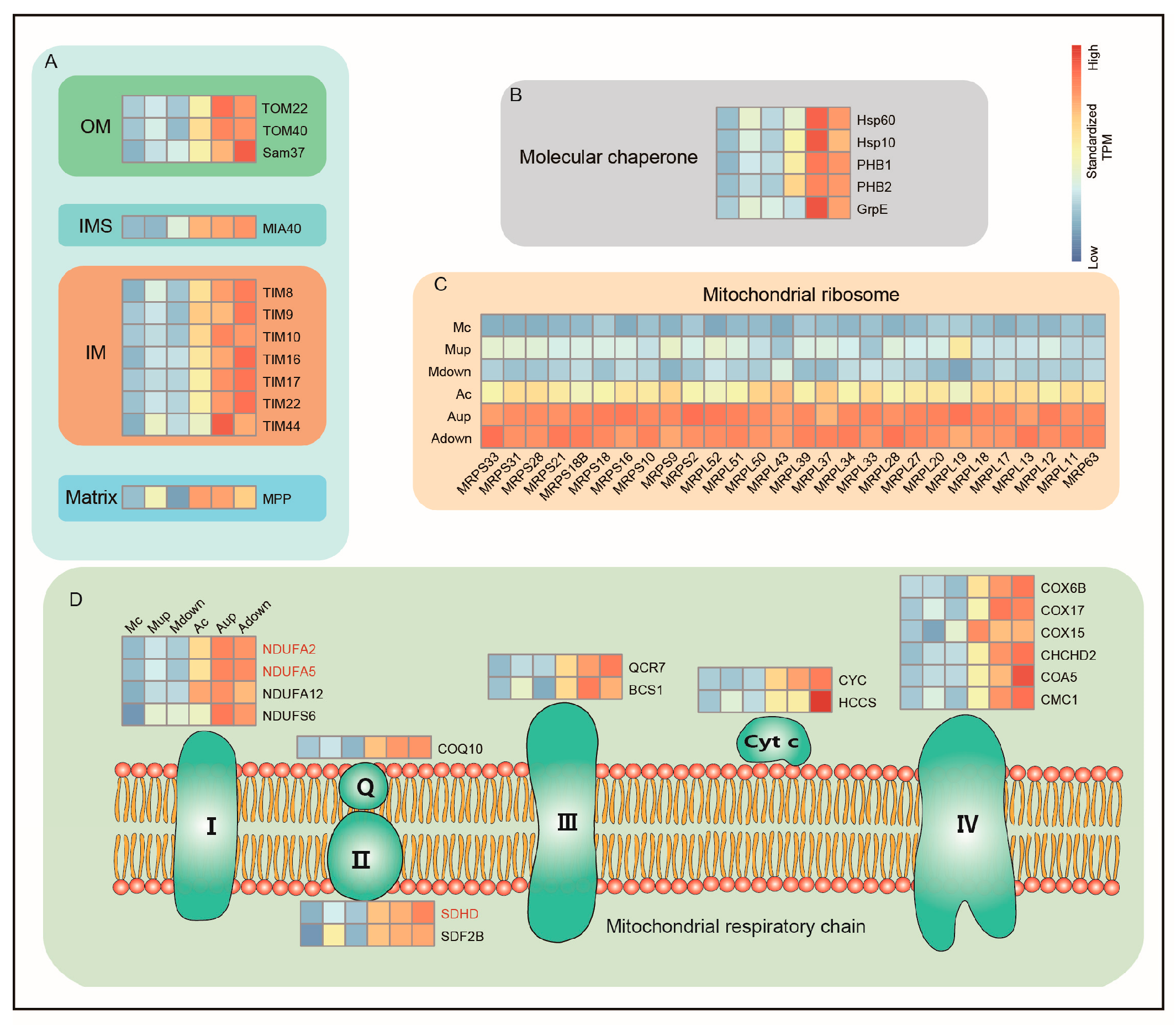
3.3. Artificial Diets Activate Mitochondrial Function in the Malpighian Tubules of Silkworms
4. Materials and Methods
4.1. Insects and Diets
4.2. Insect Rearing
4.3. Developmental Parameters Measurement and Sample Collection
4.4. RNA Preparation and Transcriptome Sequencing
4.5. Transcriptome Data Analysis
4.6. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.7. Measurement of Metabolites
4.8. Metabolome Data Analysis
4.9. GST and CYP Activity Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.J.; Zhang, S.S.; Niu, B.L.; Ji, D.F.; Liu, X.J.; Li, M.W.; Bai, H.; Palli, S.R.; Wang, C.Z.; Tan, A.J. A determining factor for insect feeding preference in the silkworm, Bombyx mori. PLoS Biol. 2019, 17, e3000162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashiya, K.; Naito, K.I.; Nishida, J.; Hamamura, Y. Silkworm rearing on artificial diet. Nippon. Nōgeikagaku Kaishi 2001, 37, 735–737. [Google Scholar] [CrossRef] [Green Version]
- Qin, D.; Wang, G.; Dong, Z.; Xia, Q.; Zhao, P. Comparative Fecal Metabolomes of Silkworms Being Fed Mulberry Leaf and Artificial Diet. Insects 2020, 11, 851. [Google Scholar] [CrossRef]
- Kiron, V.; Park, Y.; Siriyappagouder, P.; Dahle, D.; Vasanth, G.K.; Dias, J.; Fernandes, J.M.O.; Sorensen, M.; Trichet, V.V. Intestinal Transcriptome Analysis Reveals Soy Derivative-Linked Changes in Atlantic Salmon. Front. Immunol. 2020, 11, 596514. [Google Scholar] [CrossRef]
- Rostagno, M.A.; Villares, A.; Guillamon, E.; Garcia-Lafuente, A.; Martinez, J.A. Sample preparation for the analysis of isoflavones from soybeans and soy foods. J. Chromatogr. A 2009, 1216, 2–29. [Google Scholar] [CrossRef]
- Bond, D.A. In vitro digestibility of the testa in tannin-free field beans (Vicia faba L.). J. Agric. Sci. 1976, 86, 561–566. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, Y.; He, J.; Yu, J.; Mao, X.; Zheng, P.; Luo, Y.; Luo, J.; Huang, Z.; Yu, B.; et al. Effects of soybean raffinose on growth performance, digestibility, humoral immunity and intestinal morphology of growing pigs. Anim. Nutr. 2021, 7, 393–399. [Google Scholar] [CrossRef]
- Yin, B.; Liu, H.; Tan, B.; Dong, X.; Chi, S.; Yang, Q.; Zhang, S. MHC II-PI(3)K/Akt/mTOR Signaling Pathway Regulates Intestinal Immune Response Induced by Soy Glycinin in Hybrid Grouper: Protective Effects of Sodium Butyrate. Front. Immunol. 2020, 11, 615980. [Google Scholar] [CrossRef]
- Despres, L.; David, J.P.; Gallet, C. The evolutionary ecology of insect resistance to plant chemicals. Trends Ecol. Evol. 2007, 22, 298–307. [Google Scholar] [CrossRef]
- Chapman, R.F. Contact chemoreception in feeding by phytophagous insects. Annu. Rev. Entomol. 2003, 48, 455–484. [Google Scholar] [CrossRef]
- Zagrobelny, M.; Bak, S.; Rasmussen, A.V.; Jorgensen, B.; Naumann, C.M.; Lindberg Moller, B. Cyanogenic glucosides and plant-insect interactions. Phytochemistry 2004, 65, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, M.R. Postgenomic chemical ecology: From genetic code to ecological interactions. J. Chem. Ecol. 2002, 28, 873–896. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.C.; d’Alencon, E.; Guy, E.; Jacquin-Joly, E.; Jaquiery, J.; Nouhaud, P.; Peccoud, J.; Sugio, A.; Streiff, R. Genomics of adaptation to host-plants in herbivorous insects. Brief Funct. Genom. 2015, 14, 413–423. [Google Scholar] [CrossRef]
- Bajda, S.; Dermauw, W.; Greenhalgh, R.; Nauen, R.; Tirry, L.; Clark, R.M.; Van Leeuwen, T. Transcriptome profiling of a spirodiclofen susceptible and resistant strain of the European red mite Panonychus ulmi using strand-specific RNA-seq. BMC Genom. 2015, 16, 974. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.A.; Hopkins, T.L. β-Glucosylation of plant phenolics by phenol β-glucosyltransferase in larval tissues of the tobacco hornworm, Manduca sexta (L.). Insect Biochem. Mol. Biol. 1993, 23, 581–589. [Google Scholar] [CrossRef]
- Luque, T.; Okano, K.; O’Reilly, D.R. Characterization of a novel silkworm (Bombyx mori) phenol UDP-glucosyltransferase. Eur. J. Biochem. 2002, 269, 819–825. [Google Scholar] [CrossRef]
- Labbe, R.; Caveney, S.; Donly, C. Genetic analysis of the xenobiotic resistance-associated ABC gene subfamilies of the Lepidoptera. Insect. Mol. Biol. 2011, 20, 243–256. [Google Scholar] [CrossRef]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Jasmonate and salicylate induce expression of herbivore cytochrome P450 genes. Nature 2002, 419, 712–715. [Google Scholar] [CrossRef]
- Francis, F.; Vanhaelen, N.; Haubruge, E. Glutathione S-transferases in the adaptation to plant secondary metabolites in the Myzus persicae aphid. Arch. Insect. Biochem. Physiol. 2005, 58, 166–174. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, S.; Liu, Q.; Wang, Y.; Wang, H.; Liu, X.; Mu, Z. An Overviewon Research and Commercialization of Artificial Diet for Silkworm (Bombyx mori) in China. Sci. Seric. 2016, 42, 3–15. [Google Scholar]
- Ito, T. Application of Artificial Diets in Sericulture. JARQ—Jpn. Agr. Res. Q 1980, 14, 163–168. [Google Scholar]
- Xiang, H.; Liu, X.; Li, M.; Zhu, Y.; Wang, L.; Cui, Y.; Liu, L.; Fang, G.; Qian, H.; Xu, A.; et al. The evolutionary road from wild moth to domestic silkworm. Nat. Ecol. Evol. 2018, 2, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yu, H.; Shen, Y.; Banno, Y.; Xiang, Z.; Zhang, Z. Phylogeny and evolutionary history of the silkworm. Sci. China Life Sci. 2012, 55, 483–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Imai, T. Cocoon quality and physical properties of the cocoon filaments produced by silkworms reared on mulberry leaves and on an artificial diet. J. Insect Biotechnol. Sericology 2010, 55, 112–117. [Google Scholar]
- Qin, L.; Qi, J.; Shen, G.; Qin, D.; Wu, J.; Song, Y.; Cao, Y.; Zhao, P.; Xia, Q. Effects of Microbial Transfer during Food-Gut-Feces Circulation on the Health of Bombyx mori. Microbiol. Spectr. 2022, 10, e0235722. [Google Scholar] [CrossRef]
- Farina, P.; Bedini, S.; Conti, B. Multiple Functions of Malpighian Tubules in Insects: A Review. Insects 2022, 13, 1001. [Google Scholar] [CrossRef]
- Zhong, X.W.; Zou, Y.; Liu, S.P.; Yi, Q.Y.; Hu, C.M.; Wang, C.; Xia, Q.Y.; Zhao, P. Proteomic-based insight into Malpighian tubules of silkworm Bombyx mori. PloS ONE 2013, 8, e75731. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kean, L.; Yang, J.; Allan, A.K.; Davies, S.A.; Herzyk, P.; Dow, J.A. Function-informed transcriptome analysis of Drosophila renal tubule. Genome Biol. 2004, 5, R69. [Google Scholar] [CrossRef] [Green Version]
- Beyenbach, K.W.; Skaer, H.; Dow, J.A. The developmental, molecular, and transport biology of Malpighian tubules. Annu. Rev. Entomol. 2010, 55, 351–374. [Google Scholar] [CrossRef]
- Dow, J.A.; Davies, S.A. The Malpighian tubule: Rapid insights from post-genomic biology. J. Insect Physiol. 2006, 52, 365–378. [Google Scholar] [CrossRef]
- Ahn, S.J.; Badenes-Perez, F.R.; Reichelt, M.; Svatos, A.; Schneider, B.; Gershenzon, J.; Heckel, D.G. Metabolic detoxification of capsaicin by UDP-glycosyltransferase in three Helicoverpa species. Arch. Insect Biochem. Physiol. 2011, 78, 104–118. [Google Scholar] [CrossRef]
- du Rand, E.E.; Human, H.; Smit, S.; Beukes, M.; Apostolides, Z.; Nicolson, S.W.; Pirk, C.W. Proteomic and metabolomic analysis reveals rapid and extensive nicotine detoxification ability in honey bee larvae. Insect Biochem. Mol. Biol. 2017, 82, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, J.; Zhu, Y.; Duan, J.; Yu, Q.; Zhang, G.; Wan, F.; Xiang, Z.H. Genome-wide analysis of cytochrome P450 monooxygenase genes in the silkworm, Bombyx mori. Gene 2011, 480, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.Y.; Lu, C.; Li, W.L.; Xiang, Z.H.; Zhang, Z. Annotation and expression of carboxylesterases in the silkworm, Bombyx mori. BMC Genom. 2009, 10, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Lu, C.; Li, B.; Fang, S.; Zuo, W.; Dai, F.; Zhang, Z.; Xiang, Z. Identification, genomic organization and expression pattern of glutathione S-transferase in the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2008, 38, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Vogel, H.; Heckel, D.G. Comparative analysis of the UDP-glycosyltransferase multigene family in insects. Insect Biochem. Mol. Biol. 2012, 42, 133–147. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, S.; Tian, L.; Guo, E.; Luan, Y.; Zhang, J.; Li, S. Genome-wide identification and characterization of ATP-binding cassette transporters in the silkworm, Bombyx mori. BMC Genom. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Joost, H.G.; Thorens, B. The extended GLUT-family of sugar/polyol transport facilitators: Nomenclature, sequence characteristics, and potential function of its novel members (review). Mol. Membr. Biol. 2001, 18, 247–256. [Google Scholar] [CrossRef]
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Asp. Med. 2013, 34, 413–435. [Google Scholar] [CrossRef]
- Takagishi, T.; Hara, T.; Fukada, T. Recent Advances in the Role of SLC39A/ZIP Zinc Transporters In Vivo. Int. J. Mol. Sci. 2017, 18, 2708. [Google Scholar] [CrossRef] [Green Version]
- Stout, K.A.; Dunn, A.R.; Hoffman, C.; Miller, G.W. The Synaptic Vesicle Glycoprotein 2: Structure, Function, and Disease Relevance. ACS Chem. Neurosci. 2019, 10, 3927–3938. [Google Scholar] [CrossRef]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—protecting organelle protein homeostasis. J. Cell Sci. 2010, 123 Pt 22, 3849–3855. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Kiparaki, M.; Blanco, J.; Folgado, V.; Ji, Z.; Kumar, A.; Rimesso, G.; Baker, N.E. A Regulatory Response to Ribosomal Protein Mutations Controls Translation, Growth, and Cell Competition. Dev. Cell 2018, 46, 456–469.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, A.; Lingutla, R.; Mager, J. Expression analysis of mammalian mitochondrial ribosomal protein genes. Gene Expr. Patterns 2020, 38, 119147. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.A. The role of cytochrome P450 monooxygenases in plant-insect interactions. Plant Physiol 1996, 112, 1411–1419. [Google Scholar] [CrossRef] [Green Version]
- Fogleman, J.C.; Danielson, P.B.; MacIntyre, R.J. The molecular basis of adaptation in Drosophila—The role of cytochrome P450s. Evol. Biol. 1998, 30, 15–77. [Google Scholar]
- Wadleigh, R.W.; Yu, S.J. Glutathione transferase activity of fall armyworm larvae toward α,β-unsaturated carbonyl allelochemicals and its induction by allelochemicals. Insect Biochem. 1987, 17, 759–764. [Google Scholar] [CrossRef]
- Chahine, S.; O’Donnell, M.J. Interactions between detoxification mechanisms and excretion in Malpighian tubules of Drosophila melanogaster. J. Exp. Biol. 2011, 214 Pt 3, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Ling, J.; Li, M.; Mei, L. New deafness gene: Progress of research on ABCC1 in biological barriers. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2021, 38, 907–911. [Google Scholar]
- Ku, Y.S.; Contador, C.A.; Ng, M.S.; Yu, J.; Chung, G.; Lam, H.M. The Effects of Domestication on Secondary Metabolite Composition in Legumes. Front. Genet. 2020, 11, 581357. [Google Scholar] [CrossRef] [PubMed]
- Wink, M. Evolution of secondary metabolites in legumes (Fabaceae). S. Afr. J. Bot. 2013, 89, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Li, Y.; Zhang, Y.; Huang, J.; Liu, J.; Lin, Z.; Yu, Q.; Wu, A.; Wang, B. Widely Targeted Metabolomics Analysis Reveals Key Quality-Related Metabolites in Kernels of Sweet Corn. Int. J. Genom. 2021, 2021, 2654546. [Google Scholar] [CrossRef] [PubMed]
- Zurita, J.L.; Jos, A.; del Peso, A.; Salguero, M.; Lopez-Artiguez, M.; Repetto, G. Ecotoxicological effects of the antioxidant additive propyl gallate in five aquatic systems. Water Res. 2007, 41, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Becker, L. Final report on the amended safety assessment of Propyl Gallate. Int. J. Toxicol. 2007, 26, 89–118. [Google Scholar]
- Wu, X.; Chen, X.; Ye, A.; Cao, J.; He, R.; Pan, M.; Jin, F.; Ma, H.; Zhou, W. Multi-tissue metabolomic profiling reveals potential mechanisms of cocoon yield in silkworms (Bombyx mori) fed formula feed versus mulberry leaves. Front. Mol. Biosci. 2022, 9, 977047. [Google Scholar] [CrossRef]
- Xin, S.; Zhang, W. Construction and analysis of the protein-protein interaction network for the olfactory system of the silkworm Bombyx mori. Arch. Insect Biochem. Physiol. 2020, 105, e21737. [Google Scholar] [CrossRef]
- Hoglund, P.J.; Nordstrom, K.J.; Schioth, H.B.; Fredriksson, R. The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol. Biol. Evol. 2011, 28, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Doege, H.; Schurmann, A.; Bahrenberg, G.; Brauers, A.; Joost, H.G. GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. J. Biol. Chem. 2000, 275, 16275–16280. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, M.; Susztak, K.; Ranalletta, M.; Raff, A.C.; Bottinger, E.P.; Charron, M.J. Localization of the GLUT8 glucose transporter in murine kidney and regulation in vivo in nondiabetic and diabetic conditions. Am J Physiol Ren. Physiol 2005, 289, F186–F193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Qin, Q.; Zhou, B. Functional studies of Drosophila zinc transporters reveal the mechanism for zinc excretion in Malpighian tubules. BMC Biol. 2017, 15, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wu, A.; Zhang, Z.; Yan, G.; Zhang, F.; Zhang, L.; Shen, X.; Hu, R.; Zhang, Y.; Zhang, K.; et al. Characterization of the GufA subfamily member SLC39A11/Zip11 as a zinc transporter. J. Nutr. Biochem. 2013, 24, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; O’Malley, K.E.; Perry, E.A.; Balsa, E.; Latorre-Muro, P.; Riley, C.L.; Luo, C.; Jedrychowski, M.; Gygi, S.P.; Puigserver, P. Peroxisomal-derived ether phospholipids link nucleotides to respirasome assembly. Nat. Chem. Biol. 2021, 17, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Huttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Huttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Greber, B.J.; Ban, N. Structure and Function of the Mitochondrial Ribosome. Annu. Rev. Biochem. 2016, 85, 103–132. [Google Scholar] [CrossRef]
- Kutik, S.; Guiard, B.; Meyer, H.E.; Wiedemann, N.; Pfanner, N. Cooperation of translocase complexes in mitochondrial protein import. J. Cell Biol. 2007, 179, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef]
- Bo, L.; Dewey, C.N.J.B.B.; Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramirez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Metabolite | VIP | FC(A/M) | p-Value |
|---|---|---|---|---|
| Benzoic acids and derivatives | N-Propyl gallate | 2.8115 | 564.9008 | 2.64 × 10−14 |
| Carbonyl compounds | 3-Hydroxy-2-(4-methylbenzoyl)-4H-1-benzopyran-4-one | 2.0746 | 110.4676 | 6.74 × 10−13 |
| 2′,3′,4′-Trihydroxyacetophenone | 1.9507 | 3.5929 | 4.06 × 10−8 | |
| terpenoids | 3beta-3-Hydroxy-18-lupen-21-one | 2.7671 | 104.2965 | 1.17 × 10−10 |
| Shyobunyl acetate | 2.4734 | 10.115 | 1.64 × 10−11 | |
| Cincassiol B | 2.2756 | 7.2251 | 7.75 × 10−12 | |
| 4,5-Dihydrovomifoliol | 2.2691 | 4.1957 | 2.31 × 10−7 | |
| Kessyl glycol | 2.1987 | 2.9575 | 1.66 × 10−11 | |
| Acorusnol | 1.4963 | 2.0766 | 8.03 × 10−5 | |
| Organic acids and derivatives | N-[2-(3,4-dimethoxyphenyl)ethyl]-3-[4-methoxy-3-(sulfooxy)phenyl]prop-2-enimidic acid | 2.4091 | 71.6817 | 4.78 × 10−14 |
| 2-Methylcitric acid | 2.4296 | 20.3789 | 7.10 × 10−14 | |
| lipids | 7b-Hydroxy-3-oxo-5b-cholanoic acid | 2.2763 | 31.4158 | 3.04 × 10−8 |
| Heptadecanoyl carnitine | 2.2827 | 8.0294 | 1.45 × 10−13 | |
| 19-Norandrosterone | 2.0716 | 7.7129 | 2.89 × 10−10 | |
| 13-Oxo-9,11-tridecadienoic acid | 2.115 | 5.1548 | 3.97 × 10−9 | |
| 5,6-Dihydroxyprostaglandin F1a | 2.1642 | 4.8017 | 4.49 × 10−8 | |
| Hexadecanedioic acid | 1.6981 | 3.3842 | 1.02 × 10−5 | |
| Prostaglandin E2 | 1.9552 | 3.0415 | 1.33 × 10−11 | |
| Carbohydrates and carbohydrate conjugates | [6]-Gingerdiol 4′-O-beta-D-glucopyranoside | 1.9581 | 14.7255 | 6.27 × 10−12 |
| Domesticoside | 2.071 | 5.9443 | 3.48 × 10−9 | |
| Lacosamide-glucuronide | 1.5549 | 2.588 | 8.18 × 10−7 | |
| flavonoids | Cycloalliin | 2.5788 | 7.4799 | 2.38 × 10−16 |
| Austalide K | 1.9494 | 2.583 | 2.55 × 10−10 | |
| Pelargonidin 3-O-glucoside | 1.8203 | 2.3313 | 1.38 × 10−7 | |
| Apigetrin | 2.3895 | 2.0253 | 4.76 × 10−13 | |
| amino acids and their derivatives | Lysyl-Lysine | 1.9195 | 7.6448 | 1.51 × 10−8 |
| Kinetensin 4-7 | 1.9371 | 4.1428 | 1.96 × 10−12 | |
| Phenylalanyl-Arginine | 1.8862 | 2.721 | 3.52 × 10−9 | |
| Alkaloids | 3-Carboxy-1-hydroxypropylthiamine diphosphate | 2.2551 | 4.7787 | 1.28 × 10−9 |
| 1-(1,2,3,4,5-Pentahydroxypent-1-yl)-1,2,3,4-tetrahydro-beta-carboline-3-carboxylate | 1.8015 | 4.5259 | 4.51 × 10−9 | |
| Terpene glycosides | Glucosyl (2E,6E,10x)-10,11-dihydroxy-2,6-farnesadienoate | 1.8158 | 4.5214 | 2.52 × 10−8 |
| Tsangane L 3-glucoside | 1.6478 | 2.5489 | 2.65 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Zhao, D.; Wang, G.; He, Q.; Song, Y.; Jiang, Y.; Xia, Q.; Zhao, P. Adaptive Changes in Detoxification Metabolism and Transmembrane Transport of Bombyx mori Malpighian Tubules to Artificial Diet. Int. J. Mol. Sci. 2023, 24, 9949. https://doi.org/10.3390/ijms24129949
Liu L, Zhao D, Wang G, He Q, Song Y, Jiang Y, Xia Q, Zhao P. Adaptive Changes in Detoxification Metabolism and Transmembrane Transport of Bombyx mori Malpighian Tubules to Artificial Diet. International Journal of Molecular Sciences. 2023; 24(12):9949. https://doi.org/10.3390/ijms24129949
Chicago/Turabian StyleLiu, Lijing, Dongchao Zhao, Genhong Wang, Qingxiu He, Yuwei Song, Yulu Jiang, Qingyou Xia, and Ping Zhao. 2023. "Adaptive Changes in Detoxification Metabolism and Transmembrane Transport of Bombyx mori Malpighian Tubules to Artificial Diet" International Journal of Molecular Sciences 24, no. 12: 9949. https://doi.org/10.3390/ijms24129949