
Secretogranin III Selectively Promotes Vascular Leakage in the Deep Vascular Plexus of Diabetic Retinopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
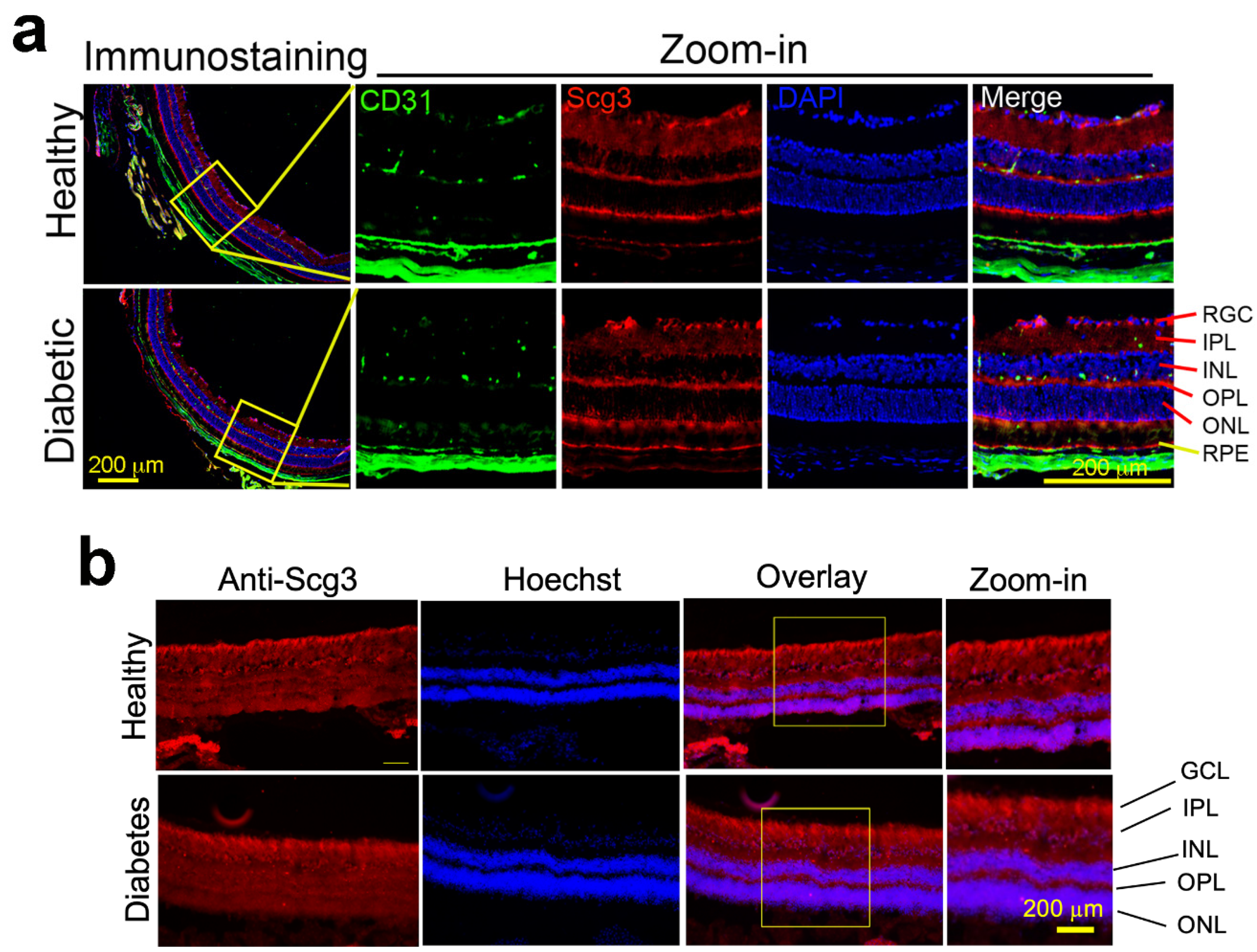
2.1. Constitutive Scg3 Expression in Diabetic and Healthy Retinas
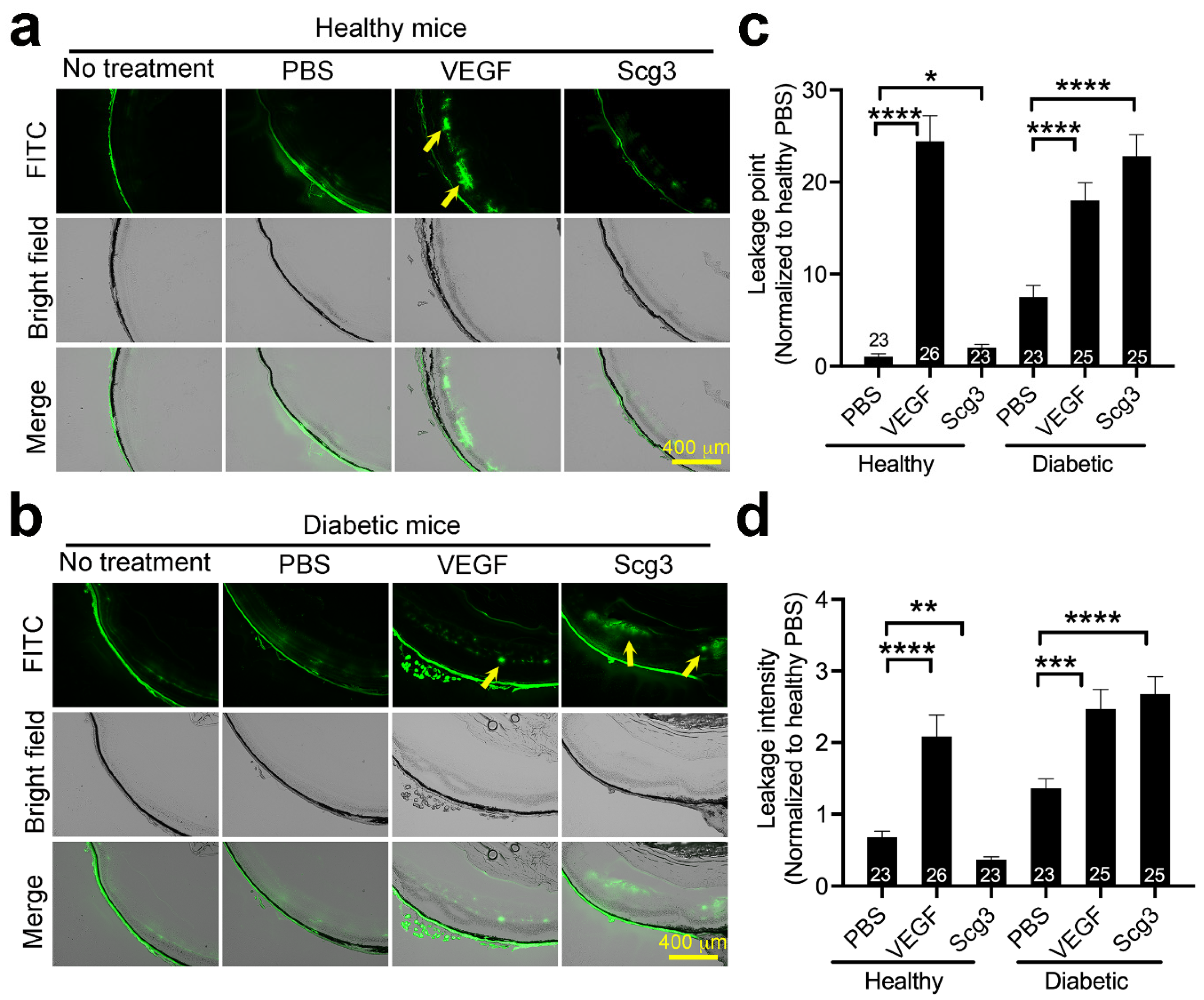
2.2. Scg3 Selectively Induces Vascular Leakage in Diabetic but Not Healthy Retinas
2.3. Scg3 Selectively Binds to Diabetic Retinas but Not Other Organs
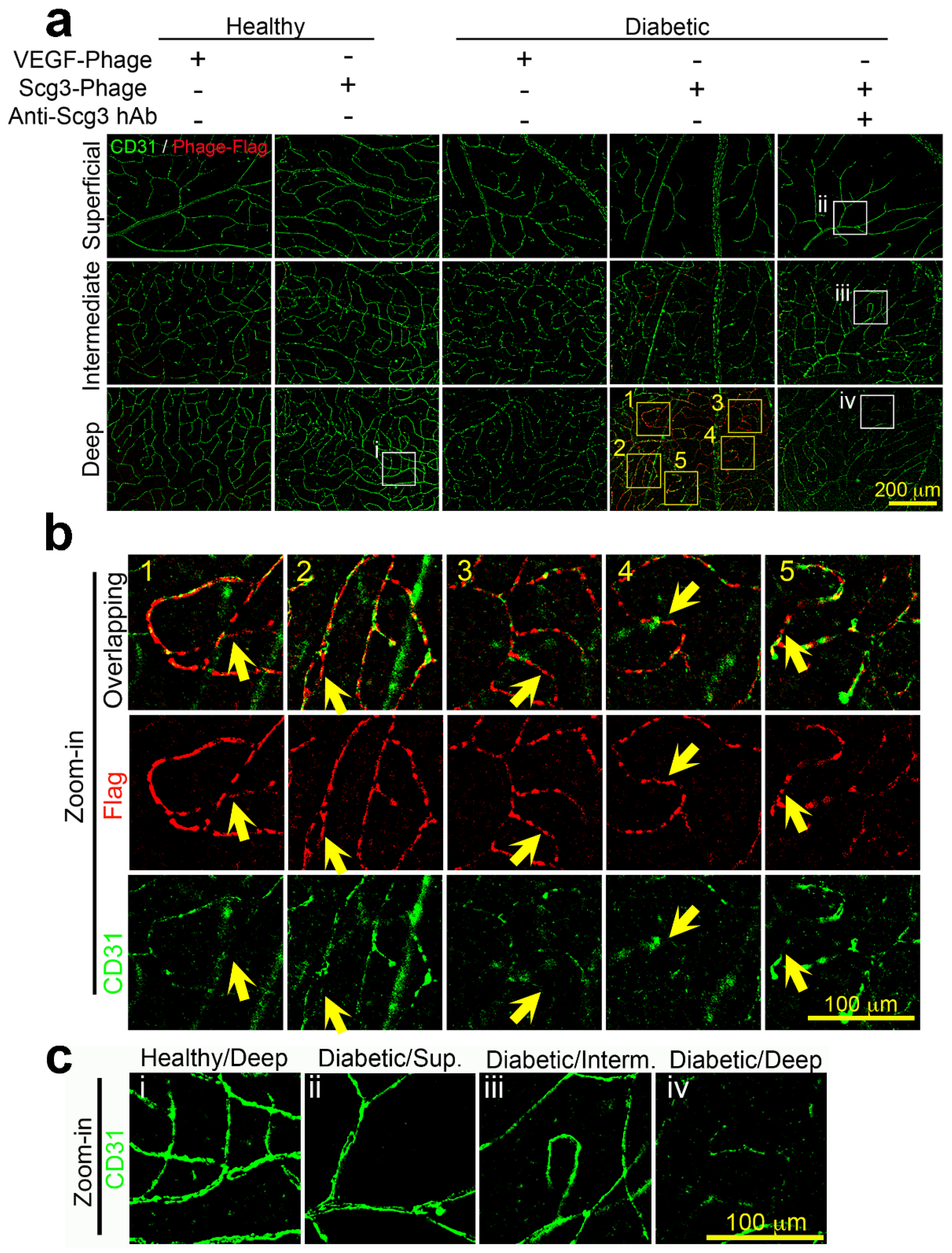
2.4. Scg3 Preferentially Binds to the Deep Vascular Plexus in Diabetic Retinas
2.5. Neutralizing Activity of anti-Scg3 hAb
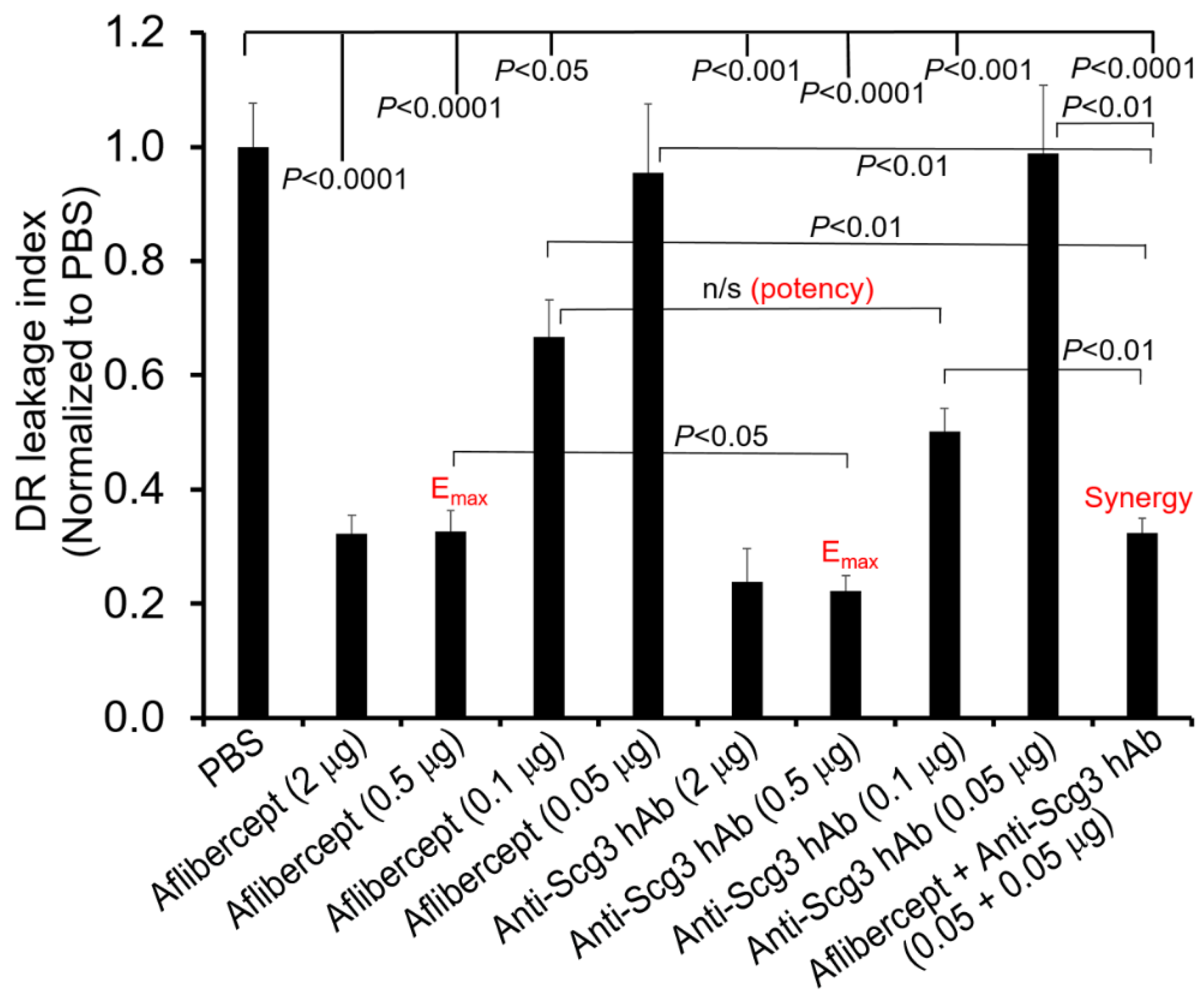
2.6. Scg3 and Aflibercept Alleviate DR Leakage with Similar High Efficacy
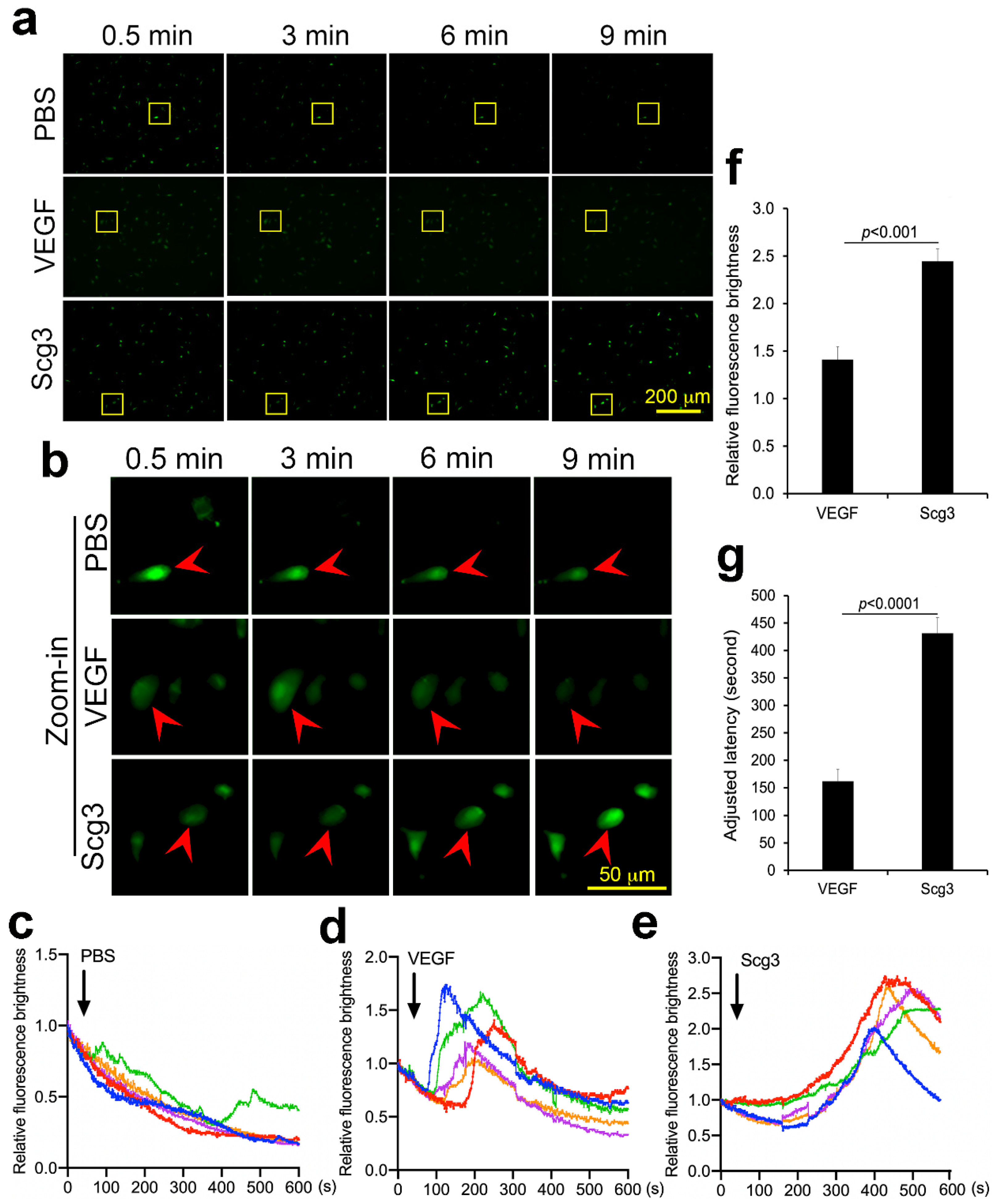
2.7. Scg3 and VEGF Differentially Induce Calcium Influx
2.8. Synergism of Anti-Scg3 and Anti-VEGF in Combination Therapy
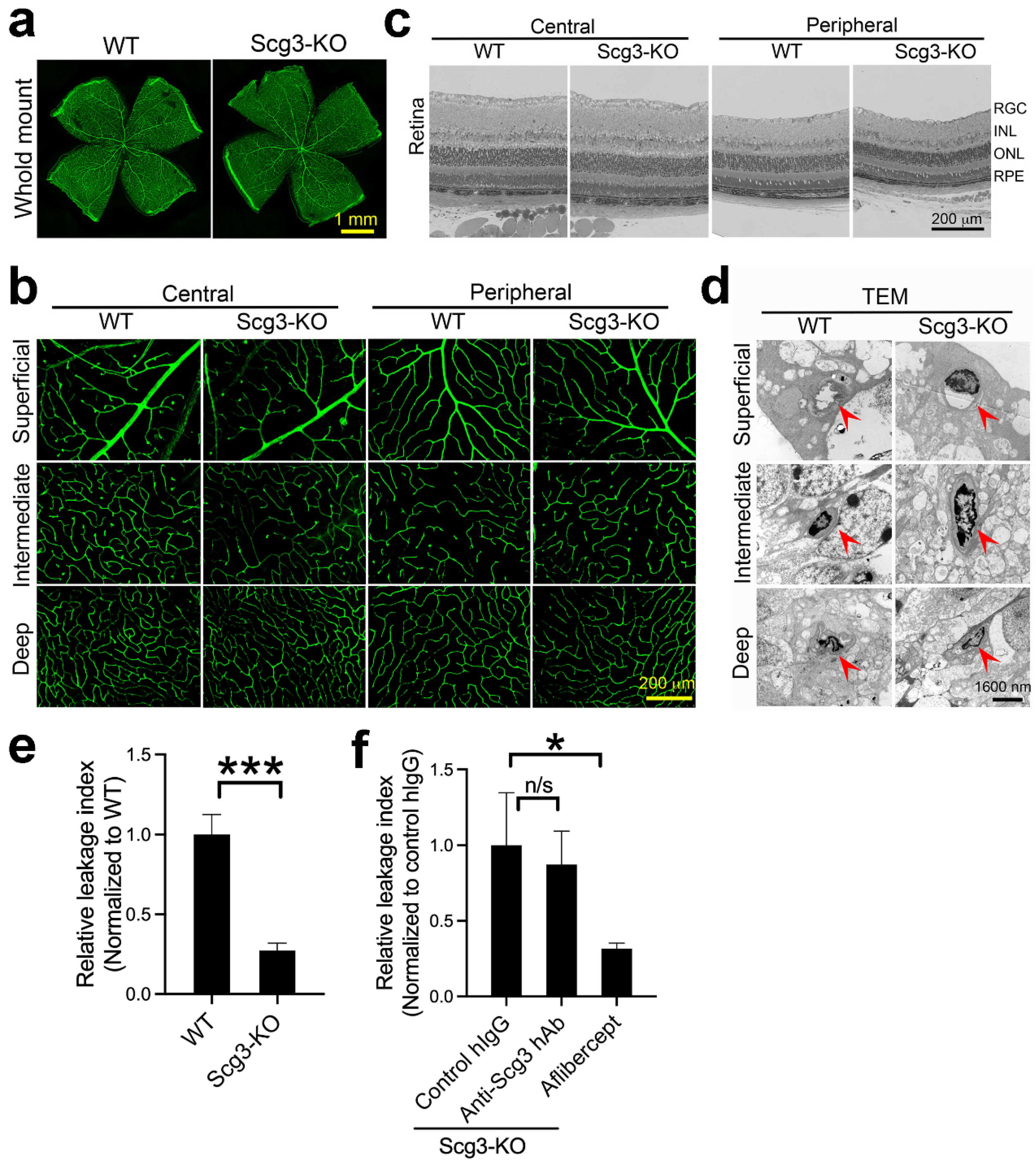
2.9. Reduced Severity of DR Leakage and Anti-Scg3 Therapy in Scg3−/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Materials
4.2. In Vivo Ligand Binding Assay
4.3. Functional Immunohistochemistry (FIHC)
4.4. Calcium Influx Assay
4.5. In Vivo FITC-dextran Leakage Assay
4.6. DR Therapy
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef] [Green Version]
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.T.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ. Res. 2005, 96, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, K.L.; Pannirselvam, M.; Anderson, T.J.; Jenkins, A.J.; Triggle, C.R.; Hill, M.A. The vascular endothelium in diabetes: A practical target for drug treatment? Expert Opin. Ther. Targets 2005, 9, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Tham, Y.-C.; Yu, M.; Chee, M.L.; Rim, T.H.; Cheung, N.; Bikbov, M.M.; Wang, Y.X.; Tang, Y.; Lu, Y.; et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-analysis. Ophthalmology 2021, 128, 1580–1591. [Google Scholar] [CrossRef]
- Ji, L.; Tian, H.; Webster, K.A.; Li, W. Neurovascular regulation in diabetic retinopathy and emerging therapies. Cell. Mol. Life Sci. CMLS 2021, 78, 5977–5985. [Google Scholar] [CrossRef]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.H.; Ma, J.-X. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H738–749. [Google Scholar] [CrossRef] [Green Version]
- Dedania, V.S.; Bakri, S.J. Current perspectives on ranibizumab. Clin. Ophthalmol. 2015, 9, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.M.; Wykoff, C.C.; Boyer, D.; Heier, J.S.; Clark, W.L.; Emanuelli, A.; Higgins, P.M.; Singer, M.; Weinreich, D.M.; Yancopoulos, G.D.; et al. Evaluation of Intravitreal Aflibercept for the Treatment of Severe Nonproliferative Diabetic Retinopathy: Results From the PANORAMA Randomized Clinical Trial. JAMA Ophthalmol. 2021, 139, 946–955. [Google Scholar] [CrossRef]
- Liu, C.-H.; Wang, Z.; Sun, Y.; Chen, J. Animal models of ocular angiogenesis: From development to pathologies. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 4665–4681. [Google Scholar] [CrossRef] [Green Version]
- Wong-Riley, M.T.T. Energy metabolism of the visual system. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, Q.V.; Linsenmeier, R.A.; Chung, C.K.; Curcio, C.A. Photoreceptor inner segments in monkey and human retina: Mitochondrial density, optics, and regional variation. Vis. Neurosci. 2002, 19, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Simonett, J.M.; Scarinci, F.; Picconi, F.; Giorno, P.; De Geronimo, D.; Di Renzo, A.; Varano, M.; Frontoni, S.; Parravano, M. Early microvascular retinal changes in optical coherence tomography angiography in patients with type 1 diabetes mellitus. Acta Ophthalmol. 2017, 95, e751–e755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waduge, P.; Tian, H.; Webster, K.A.; Li, W. Profiling disease-selective drug targets: From proteomics to ligandomics. Drug Discov. Today 2022, 28, 103430. [Google Scholar] [CrossRef]
- LeBlanc, M.E.; Wang, W.; Chen, X.; Caberoy, N.B.; Guo, F.; Shen, C.; Ji, Y.; Tian, H.; Wang, H.; Chen, R.; et al. Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J. Exp. Med. 2017, 214, 1029–1047. [Google Scholar] [CrossRef] [Green Version]
- Rong, X.; Tian, H.; Yang, L.; Li, W. Function-first ligandomics for ocular vascular research and drug target discovery. Exp. Eye Res. 2019, 182, 57–64. [Google Scholar] [CrossRef]
- Tang, F.; LeBlanc, M.E.; Wang, W.; Liang, D.; Chen, P.; Chou, T.-H.; Tian, H.; Li, W. Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis 2019, 22, 369–382. [Google Scholar] [CrossRef]
- Dai, C.; Waduge, P.; Ji, L.; Huang, C.; He, Y.; Tian, H.; Zuniga-Sanchez, E.; Bhatt, A.; Pang, I.-H.; Su, G.; et al. Secretogranin III stringently regulates pathological but not physiological angiogenesis in oxygen-induced retinopathy. Cell. Mol. Life Sci. CMLS 2022, 79, 63. [Google Scholar] [CrossRef]
- Li, W.; Pang, I.-H.; Pacheco, M.T.F.; Tian, H. Ligandomics: A paradigm shift in biological drug discovery. Drug Discov. Today 2018, 23, 636–643. [Google Scholar] [CrossRef]
- Li, J.; Wang, P.; Ying, J.; Chen, Z.; Yu, S. Curcumin Attenuates Retinal Vascular Leakage by Inhibiting Calcium/Calmodulin-Dependent Protein Kinase II Activity in Streptozotocin-Induced Diabetes. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 1196–1208. [Google Scholar] [CrossRef]
- Ji, L.; Waduge, P.; Hao, L.; Kaur, A.; Wan, W.; Wu, Y.; Li, W. Selectively targeting disease-restricted secretogranin III to alleviate choroidal neovascularization. FASEB J. 2022, 36, e22106. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, D.M.; Rinchik, E.M.; Russell, L.B.; Ottiger, H.P.; Sutcliffe, J.G.; Copeland, N.G.; Jenkins, N.A. Genetic ablation of a mouse gene expressed specifically in brain. EMBO J. 1990, 9, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Webster, K.A.; Bhatt, A.; Tian, H.; Su, G.; Li, W. Concurrent Physiological and Pathological Angiogenesis in Retinopathy of Prematurity and Emerging Therapies. Int. J. Mol. Sci. 2021, 22, 4809. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Dietenbeck, T.; Giron, A.; Paques, M.; Kachenoura, N.; Girerd, X. Non-invasive evaluation of retinal vascular remodeling and hypertrophy in humans: Intricate effect of ageing, blood pressure and glycaemia. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2021, 110, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, R.; González-Álvarez, M.; Delgado, R.; Esteban, S.; Arroyo, A.G. Remodeling of the Microvasculature: May the Blood Flow Be With You. Front. Physiol. 2020, 11, 586852. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Nittala, M.G.; Villanueva Boone, C.; Yu, H.J.; Fan, W.; Velaga, S.B.; Ehlers, J.P.; Ip, M.S.; Sadda, S.R. RECOVERY Study Group Final Outcomes from the Randomized RECOVERY Trial of Aflibercept for Retinal Nonperfusion in Proliferative Diabetic Retinopathy. Ophthalmol. Retina 2022, 6, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Lutty, G.A.; McLeod, D.S.; Bhutto, I.; Wiegand, S.J. Effect of VEGF trap on normal retinal vascular development and oxygen-induced retinopathy in the dog. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4039–4047. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, Y.; Cao, Z.; Ji, H.; Yang, X.; Iwamoto, H.; Wahlberg, E.; Länne, T.; Sun, B.; Cao, Y. Anti-VEGF- and anti-VEGF receptor-induced vascular alteration in mouse healthy tissues. Proc. Natl. Acad. Sci. USA 2013, 110, 12018–12023. [Google Scholar] [CrossRef] [Green Version]
- Elnahry, A.G.; Abdel-Kader, A.A.; Habib, A.E.; Elnahry, G.A.; Raafat, K.A.; Elrakhawy, K. Review on Recent Trials Evaluating the Effect of Intravitreal Injections of Anti-VEGF Agents on the Macular Perfusion of Diabetic Patients with Diabetic Macular Edema. Rev. Recent Clin. Trials 2020, 15, 188–198. [Google Scholar] [CrossRef]
- Abri Aghdam, K.; Reznicek, L.; Soltan Sanjari, M.; Klingenstein, A.; Kernt, M.; Seidensticker, F. Anti-VEGF treatment and peripheral retinal nonperfusion in patients with central retinal vein occlusion. Clin. Ophthalmol. 2017, 11, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Resch, M.D.; Balogh, A.; Deák, G.G.; Nagy, Z.Z.; Papp, A. Vascular density in age-related macular degeneration after one year of antiVEGF treatment with treat-and-extend and fixed regimens. PLoS ONE 2020, 15, e0229388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.-Y.; Meng, Y.-A.; Yan, B.; Luo, J. Effect of anti-VEGF treatment on nonperfusion areas in ischemic retinopathy. Int. J. Ophthalmol. 2021, 14, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Tonade, D.; Kern, T.S. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog. Retin. Eye Res. 2021, 83, 100919. [Google Scholar] [CrossRef] [PubMed]
- Nesper, P.L.; Roberts, P.K.; Onishi, A.C.; Chai, H.; Liu, L.; Jampol, L.M.; Fawzi, A.A. Quantifying Microvascular Abnormalities With Increasing Severity of Diabetic Retinopathy Using Optical Coherence Tomography Angiography. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO307–BIO315. [Google Scholar] [CrossRef]
- Agemy, S.A.; Scripsema, N.K.; Shah, C.M.; Chui, T.; Garcia, P.M.; Lee, J.G.; Gentile, R.C.; Hsiao, Y.-S.; Zhou, Q.; Ko, T.; et al. Retinal Vascular Perfusion Density Mapping Using Optical Coherence Tomography Angiography in Normals and Diabetic Retinopathy Patients. Retina 2015, 35, 2353–2363. [Google Scholar] [CrossRef]
- Tan, T.-E.; Nguyen, Q.; Chua, J.; Schmetterer, L.; Tan, G.S.W.; Wong, C.W.; Tsai, A.; Cheung, G.C.M.; Wong, T.Y.; Ting, D.S.W. Global Assessment of Retinal Arteriolar, Venular and Capillary Microcirculations Using Fundus Photographs and Optical Coherence Tomography Angiography in Diabetic Retinopathy. Sci. Rep. 2019, 9, 11751. [Google Scholar] [CrossRef] [Green Version]
- Eshaq, R.S.; Harris, N.R. Hyperglycemia-induced ubiquitination and degradation of β-catenin with the loss of platelet endothelial cell adhesion molecule-1 in retinal endothelial cells. Microcirculation 2020, 27, e12596. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Fields, M.A.; Del Priore, L.V.; Adelman, R.A.; Rizzolo, L.J. Interactions of the choroid, Bruch’s membrane, retinal pigment epithelium, and neurosensory retina collaborate to form the outer blood-retinal-barrier. Prog. Retin. Eye Res. 2020, 76, 100803. [Google Scholar] [CrossRef]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef]
- Matsunaga, N.; Chikaraishi, Y.; Izuta, H.; Ogata, N.; Shimazawa, M.; Matsumura, M.; Hara, H. Role of soluble vascular endothelial growth factor receptor-1 in the vitreous in proliferative diabetic retinopathy. Ophthalmology 2008, 115, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Fico, E.; Rosso, P.; Triaca, V.; Segatto, M.; Lambiase, A.; Tirassa, P. NGF Prevents Loss of TrkA/VEGFR2 Cells, and VEGF Isoform Dysregulation in the Retina of Adult Diabetic Rats. Cells 2022, 11, 3246. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Khanani, A.M.; Quezada Ruiz, C.; Basu, K.; Ferrone, P.J.; Brittain, C.; Figueroa, M.S.; Lin, H.; Holz, F.G.; Patel, V.; et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): Two randomised, double-masked, phase 3, non-inferiority trials. Lancet 2022, 399, 729–740. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Abreu, F.; Adamis, A.P.; Basu, K.; Eichenbaum, D.A.; Haskova, Z.; Lin, H.; Loewenstein, A.; Mohan, S.; Pearce, I.A.; et al. Efficacy, durability, and safety of intravitreal faricimab with extended dosing up to every 16 weeks in patients with diabetic macular oedema (YOSEMITE and RHINE): Two randomised, double-masked, phase 3 trials. Lancet 2022, 399, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Waduge, P.; Wan, W.; Tian, H.; Li, J.; Zhang, J.; Li, W. Comparative ligandomics implicates secretogranin III as a disease-restricted angiogenic factor in laser-induced choroidal neovascularization. FEBS J. 2022, 289, 3521–3534. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Zhou, Y.; Jiang, X.; Alvarado, G.; Li, W. Efficient identification of tubby-binding proteins by an improved system of T7 phage display. J. Mol. Recognit. 2010, 23, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, K.R.; Brown, K.A.; Chen, W.U.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and physiological characterization of fluo-4 Ca(2+)-indicator dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef]
- Xu, H.Z.; Le, Y.Z. Significance of outer blood-retina barrier breakdown in diabetes and ischemia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2160–2164. [Google Scholar] [CrossRef]
- Guo, F.; Ding, Y.; Caberoy, N.; Alvarado, G.; Wang, F.; Chen, R.; Li, W. ABCF1 extrinsically regulates retinal pigment epithelial cell phagocytosis. Mol. Biol. Cell 2015, 26, 2311–2320. [Google Scholar] [CrossRef]
- Tang, F.; Pacheco, M.T.F.; Chen, P.; Liang, D.; Li, W. Secretogranin III promotes angiogenesis through MEK/ERK signaling pathway. Biochem. Biophys. Res. Commun. 2018, 495, 781–786. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, L.; Waduge, P.; Wu, Y.; Huang, C.; Kaur, A.; Oliveira, P.; Tian, H.; Zhang, J.; Stout, J.T.; Weng, C.Y.; et al. Secretogranin III Selectively Promotes Vascular Leakage in the Deep Vascular Plexus of Diabetic Retinopathy. Int. J. Mol. Sci. 2023, 24, 10531. https://doi.org/10.3390/ijms241310531
Ji L, Waduge P, Wu Y, Huang C, Kaur A, Oliveira P, Tian H, Zhang J, Stout JT, Weng CY, et al. Secretogranin III Selectively Promotes Vascular Leakage in the Deep Vascular Plexus of Diabetic Retinopathy. International Journal of Molecular Sciences. 2023; 24(13):10531. https://doi.org/10.3390/ijms241310531
Chicago/Turabian StyleJi, Liyang, Prabuddha Waduge, Yan Wu, Chengchi Huang, Avinash Kaur, Paola Oliveira, Hong Tian, Jinsong Zhang, J. Timothy Stout, Christina Y. Weng, and et al. 2023. "Secretogranin III Selectively Promotes Vascular Leakage in the Deep Vascular Plexus of Diabetic Retinopathy" International Journal of Molecular Sciences 24, no. 13: 10531. https://doi.org/10.3390/ijms241310531