

Rotenone Blocks the Glucocerebrosidase Enzyme and Induces the Accumulation of Lysosomes and Autophagolysosomes Independently of LRRK2 Kinase in HEK-293 Cells

Abstract
:1. Introduction
2. Results
2.1. Rotenone (ROT) Inhibits Glucocerebrosidase (GCase) Activity by Mimicking the Inhibitor Conduritol-β-Epoxide (CBE) in HEK-293 Cells
2.2. Rotenone (ROT) and Conduritol-β-Epoxide (CBE) Induce Accumulation of Lysosomes but ROT Affects the Mitochondrial Membrane Potential (ΔΨm) Only in HEK-293 Cells
2.3. Rotenone (ROT) and Conduritol-β-Epoxide (CBE) Induce Both the Accumulation of Lysosomes and an Increase in Autophagolysosomes in HEK-293 Cells
2.4. Rotenone (ROT) but Not Conduritol-β-Epoxide (CBE) Induces the Oxidation of Stress Sensor Protein DJ-1 and Cleaved Caspase 3 (CC3) in HEK-293 Cells
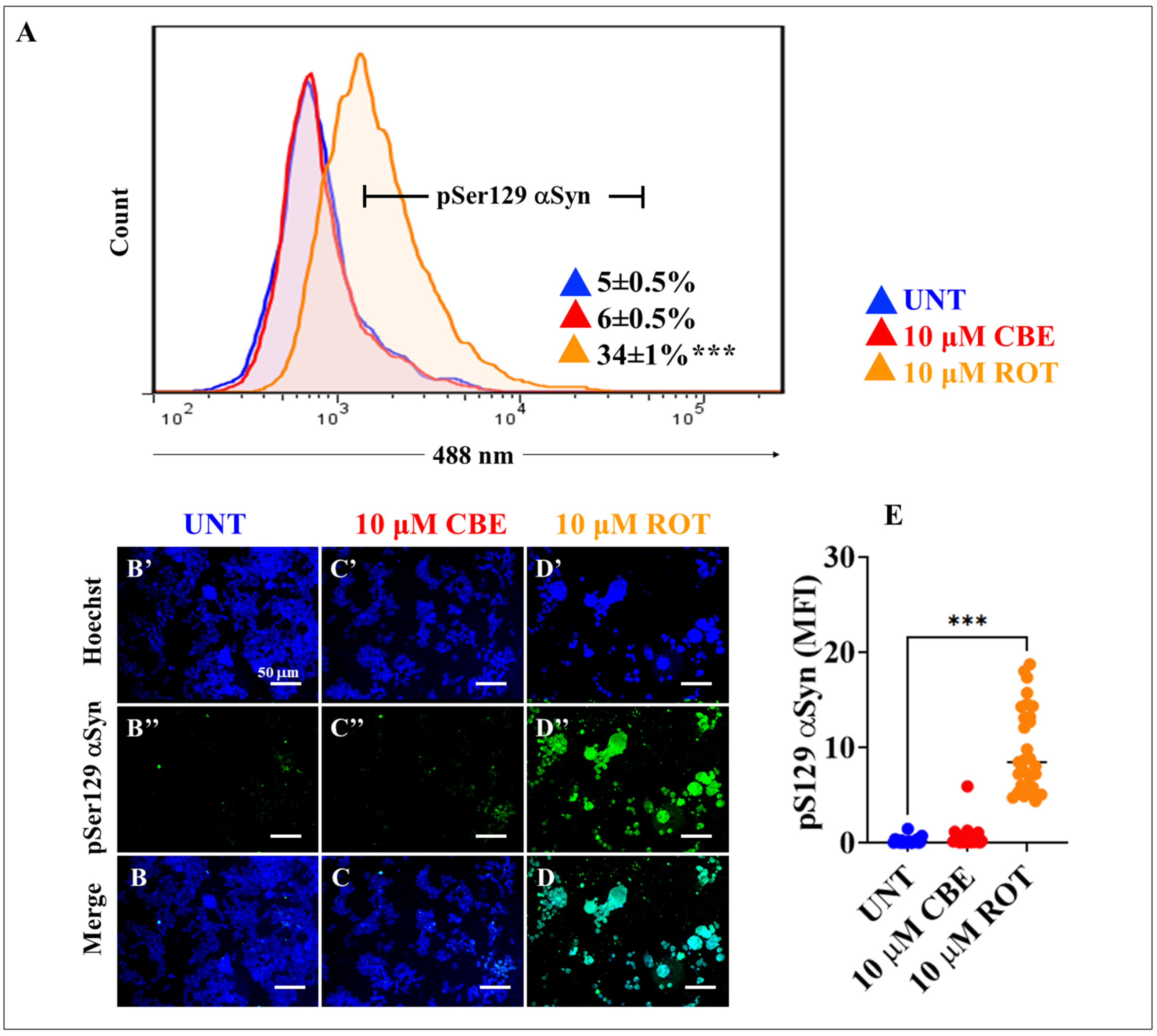
2.5. Rotenone (ROT) but Not Conduritol-β-Epoxide (CBE) Induces Phosphorylation of Alpha-synuclein (α-Syn) and LRRK2 Kinase in HEK-293 Cells
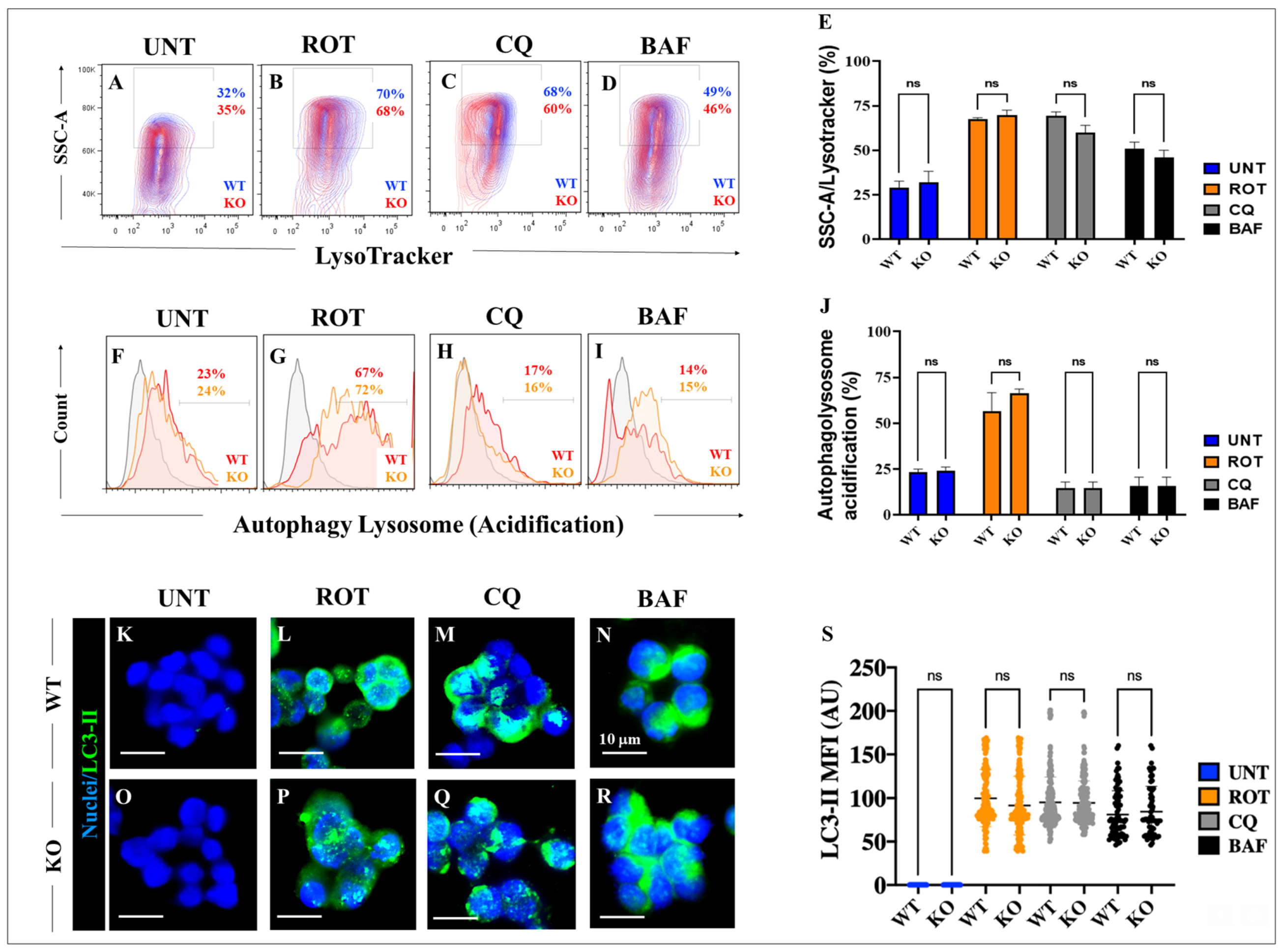
2.6. Rotenone (ROT) Does Not Induce the Phosphorylation of LRRK Kinase in HEK-293 LRRK2 Knockout (KO) Cells
2.7. ROT Inhibits the Enzymatic Activity of GCase Equally in Both WT and HEK-293 KO Cells
2.8. ROT Induces the Accumulation of Lysosomes and Reduces Mitochondrial Potential in HEK-293 KO Cells
2.9. Rotenone (ROT) Induces An Increase in Autophagosomes Independently of LRRK2
2.10. ROT Neither Induces the Phosphorylation of α-Syn, the Oxidation of DJ-1, Nor the Activation of Caspase 3 (CC3) in HEK-293 LRRK2 KO Cells
3. Discussion
4. Materials and Methods
4.1. HEK-293 Cell Line
4.2. Analysis of Cells
4.2.1. Assay Protocol
4.2.2. GCase Activity Assay
4.2.3. Characterization of Lysosomal Complexity
4.2.4. Analysis of Mitochondrial Membrane Potential (ΔΨm)
4.2.5. Detection of oxDJ-1, Cleaved Caspase 3 (CC3), LRRK2, Alpha-Synuclein, and LC3-II Using Fluorescent Microscopy and Flow Cytometry
4.2.6. Autophagy Assay
4.3. Molecular Docking
4.4. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236, discussion 222. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef]
- Lu, J.; Wu, M.; Yue, Z. Autophagy and Parkinson’s Disease. Adv. Exp. Med. Biol. 2020, 1207, 21–51. [Google Scholar] [CrossRef]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Sae-Yun, A.; Ovatlarnporn, C.; Itharat, A.; Wiwattanapatapee, R. Extraction of rotenone from Derris elliptica and Derris malaccensis by pressurized liquid extraction compared with maceration. J. Chromatogr. A 2006, 1125, 172–176. [Google Scholar] [CrossRef]
- Zubairi, S.; Sarmidi, M.; Aziz, R. A Study of Rotenone from Derris Roots of Varies Location, Plant Parts and Types of Solvent Used. Adv. Environ. Biol. 2014, 8, 445–449. [Google Scholar]
- Fang, N.; Casida, J.E. Cubé resin insecticide: Identification and biological activity of 29 rotenoid constituents. J. Agric. Food Chem. 1999, 47, 2130–2136. [Google Scholar] [CrossRef]
- Lawana, V.; Cannon, J. Chapter Five—Neurotoxicity: Relevance to Parkinson’s disease. In Advances in Neurotoxicology; Aschner, M., Costa, L., Eds.; Academic Press: New York, NY, USA, 2020; Volume 4, pp. 209–254. [Google Scholar]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Read, A.D.; Bentley, R.E.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef]
- Yahya, V.; Di Fonzo, A.; Monfrini, E. Genetic Evidence for Endolysosomal Dysfunction in Parkinson’s Disease: A Critical Overview. Int. J. Mol. Sci. 2023, 24, 6338. [Google Scholar] [CrossRef]
- Lee, C.Y.; Menozzi, E.; Chau, K.Y.; Schapira, A.H.V. Glucocerebrosidase 1 and leucine-rich repeat kinase 2 in Parkinson disease and interplay between the two genes. J. Neurochem. 2021, 159, 826–839. [Google Scholar] [CrossRef]
- Usmani, A.; Shavarebi, F.; Hiniker, A. The Cell Biology of LRRK2 in Parkinson’s Disease. Mol. Cell. Biol. 2021, 41, e00660-20. [Google Scholar] [CrossRef]
- Thirugnanam, T.; Santhakumar, K. Chemically induced models of Parkinson’s disease. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2022, 252, 109213. [Google Scholar] [CrossRef]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Neuroprotective Effect of the LRRK2 Kinase Inhibitor PF-06447475 in Human Nerve-like Differentiated Cells Exposed to Oxidative Stress Stimuli: Implications for Parkinson’s Disease. Neurochem. Res. 2016, 41, 2675–2692. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and α-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef]
- Kondo, K.; Obitsu, S.; Teshima, R. α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol. Pharm. Bull. 2011, 34, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Senkevich, K.; Gan-Or, Z. Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Park. Relat. Disord. 2020, 73, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [Green Version]
- Ellison, S.; Parker, H.; Bigger, B. Advances in therapies for neurological lysosomal storage disorders. J. Inherit. Metab. Dis. 2023. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jmoudiak, M.; Futerman, A.H. Gaucher disease: Pathological mechanisms and modern management. Br. J. Haematol. 2005, 129, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedariti, M.; Frattini, E.; Baden, P.; Cogo, S.; Civiero, L.; Ziviani, E.; Zilio, G.; Bertoli, F.; Aureli, M.; Kaganovich, A.; et al. LRRK2 kinase activity regulates GCase level and enzymatic activity differently depending on cell type in Parkinson’s disease. Npj Park. Dis. 2022, 8, 92. [Google Scholar] [CrossRef]
- Menozzi, E.; Toffoli, M.; Schapira, A.H.V. Targeting the GBA1 pathway to slow Parkinson disease: Insights into clinical aspects, pathogenic mechanisms and new therapeutic avenues. Pharmacol. Ther. 2023, 246, 108419. [Google Scholar] [CrossRef]
- Zhang, J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biol. 2015, 4, 242–259. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Hassan, A.; Lee, C.H.; Xie, X.S.; Li, X. Molecular basis of V-ATPase inhibition by bafilomycin A1. Nat. Commun. 2021, 12, 1782. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [CrossRef]
- Mader, B.J.; Pivtoraiko, V.N.; Flippo, H.M.; Klocke, B.J.; Roth, K.A.; Mangieri, L.R.; Shacka, J.J. Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem. Neurosci. 2012, 3, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Premkumar, L.; Sawkar, A.R.; Boldin-Adamsky, S.; Toker, L.; Silman, I.; Kelly, J.W.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase covalently bound to conduritol-B-epoxide. Implications for Gaucher disease. J. Biol. Chem. 2005, 280, 23815–23819. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, N.J.; Brady, R.O.; Tappel, A.L. The lysosomal localization of sphingolipid hydrolases. Biochim. Biophys. Acta 1968, 159, 141–146. [Google Scholar] [CrossRef]
- Musiwaro, P.; Smith, M.; Manifava, M.; Walker, S.A.; Ktistakis, N.T. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 2013, 9, 1407–1417. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Kinumi, T.; Kimata, J.; Taira, T.; Ariga, H.; Niki, E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 317, 722–728. [Google Scholar] [CrossRef]
- Legler, G. Glycoside hydrolases: Mechanistic information from studies with reversible and irreversible inhibitors. Adv. Carbohydr. Chem. Biochem. 1990, 48, 319–384. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Schiller, J.; Zickermann, V. Binding of Natural Inhibitors to Respiratory Complex I. Pharmaceuticals 2022, 15, 1088. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. DJ-1 as a Biomarker of Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 1037, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Oxidized DJ-1 as a possible biomarker of Parkinson’s disease. J. Clin. Biochem. Nutr. 2014, 54, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Kamikawaji, S.; Ito, G.; Iwatsubo, T. Identification of the autophosphorylation sites of LRRK2. Biochemistry 2009, 48, 10963–10975. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Moore, D.J.; Xiong, Y.; Dawson, T.M.; Dawson, V.L. Reevaluation of phosphorylation sites in the Parkinson disease-associated leucine-rich repeat kinase 2. J. Biol. Chem. 2010, 285, 29569–29576. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.; Inesta-Vaquera, F.; Zhang, J.; Xie, C.; Cai, H.; Arthur, S.; Tan, L.; Choi, H.; Gray, N.; Cohen, P.; et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS ONE 2012, 7, e39132. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Li, T.; Liu, Z.; Arbez, N.; Yan, J.; Moran, T.H.; Ross, C.A.; Smith, W.W. LRRK2 kinase activity mediates toxic interactions between genetic mutation and oxidative stress in a Drosophila model: Suppression by curcumin. Neurobiol. Dis. 2012, 47, 385–392. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeles, D.C.; Gan, B.H.; Onstead, L.; Zhao, Y.; Lim, K.L.; Dachsel, J.; Melrose, H.; Farrer, M.; Wszolek, Z.K.; Dickson, D.W.; et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 2011, 32, 1390–1397. [Google Scholar] [CrossRef]
- Yoon, J.H.; Mo, J.S.; Kim, M.Y.; Ann, E.J.; Ahn, J.S.; Jo, E.H.; Lee, H.J.; Lee, Y.C.; Seol, W.; Yarmoluk, S.M.; et al. LRRK2 functions as a scaffolding kinase of ASK1-mediated neuronal cell death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2356–2368. [Google Scholar] [CrossRef]
- Chen, C.Y.; Weng, Y.H.; Chien, K.Y.; Lin, K.J.; Yeh, T.H.; Cheng, Y.P.; Lu, C.S.; Wang, H.L. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012, 19, 1623–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.H.; Kim, H.; Kim, J.; Sim, H.; Ahn, H.; Seo, H.; Chung, K.C.; Park, B.J.; Son, I.; Seol, W. Leucine-Rich Repeat Kinase 2 (LRRK2) phosphorylates p53 and induces p21(WAF1/CIP1) expression. Mol. Brain 2015, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Quintero-Espinosa, D.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Knockdown transgenic Lrrk Drosophila resists paraquat-induced locomotor impairment and neurodegeneration: A therapeutic strategy for Parkinson’s disease. Brain Res. 2017, 1657, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Wong, W.; McGeer, E.G.; McGeer, P.L. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 2009, 387, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Akther, M.; Azam, S.; Kim, I.S.; Lin, Y.; Lee, Y.H.; Choi, D.K. Targeting α-synuclein aggregation and its role in mitochondrial dysfunction in Parkinson’s disease. Br. J. Pharmacol. 2022, 179, 23–45. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, G.; Paciotti, S.; Gatticchi, L.; Parnetti, L. The vicious cycle between α-synuclein aggregation and autophagic-lysosomal dysfunction. Mov. Disord. 2020, 35, 34–44. [Google Scholar] [CrossRef]
- Dermentzaki, G.; Dimitriou, E.; Xilouri, M.; Michelakakis, H.; Stefanis, L. Loss of β-glucocerebrosidase activity does not affect alpha-synuclein levels or lysosomal function in neuronal cells. PLoS ONE 2013, 8, e60674. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.L.; Velayati, A.; Sidransky, E.; Lee, J.C. Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol. Genet. Metab. 2013, 108, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Chen, M.; Mi, N.; Yang, W.; Li, X.; Wang, P.; Yin, N.; Li, Y.; Yue, F.; Chan, P.; et al. Increased oligomerization and phosphorylation of α-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol. Aging 2015, 36, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallings, R.; Connor-Robson, N.; Wade-Martins, R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum. Mol. Genet. 2019, 28, 2696–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.L.; Miller, A.D.; Freeman, T.C. Identification and characterisation of human apoptosis inducing proteins using cell-based transfection microarrays and expression analysis. BMC Genom. 2006, 7, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, I. The Parkinson disease gene LRRK2: Evolutionary and structural insights. Mol. Biol. Evol. 2006, 23, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [Green Version]
- Coku, I.; Mutez, E.; Eddarkaoui, S.; Carrier, S.; Marchand, A.; Deldycke, C.; Goveas, L.; Baille, G.; Tir, M.; Magnez, R.; et al. Functional Analyses of Two Novel LRRK2 Pathogenic Variants in Familial Parkinson’s Disease. Mov. Disord. 2022, 37, 1761–1767. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Kinkl, N.; Schumacher, A.; Braun, R.J.; O’Neill, E.; Meitinger, T.; Kolch, W.; Prokisch, H.; Ueffing, M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006, 15, 223–232. [Google Scholar] [CrossRef]
- Forkink, M.; Basit, F.; Teixeira, J.; Swarts, H.G.; Koopman, W.J.H.; Willems, P.H.G.M. Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol. 2015, 6, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef] [Green Version]
- Storch, A.; Ludolph, A.C.; Schwarz, J. HEK-293 cells expressing the human dopamine transporter are susceptible to low concentrations of 1-methyl-4-phenylpyridine (MPP+) via impairment of energy metabolism. Neurochem. Int. 1999, 35, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.C.; Jin, L.; Cao, Y.; Wang, L.Z.; Meng, F.; Zhu, X.Z. Cloning, expression, and functional analysis of human dopamine D1 receptors. Acta Pharmacol. Sin. 2005, 26, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Jolly, J.P.; Surmeier, J.D.; Mullah, B.M.; Lidow, M.S.; Bergson, C.M.; Robishaw, J.D. Differential dependence of the D1 and D5 dopamine receptors on the G protein gamma 7 subunit for activation of adenylylcyclase. J. Biol. Chem. 2001, 276, 39386–39393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, L.B.; Davanzo, G.G.; de Aguiar, C.F.; Moraes-Vieira, P.M.M. Using flow cytometry for mitochondrial assays. MethodsX 2020, 7, 100938. [Google Scholar] [CrossRef]
- Lazic, S.E.; Clarke-Williams, C.J.; Munafo, M.R. What exactly is ‘N’ in cell culture and animal experiments? PLoS Biol. 2018, 16, e2005282. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.Y.; Lo, R.C.N.; Ho, P.W.; Liu, H.F.; Chang, E.E.S.; Leung, C.T.; Malki, Y.; Choi, Z.Y.; Wong, W.Y.; Kung, M.H.; et al. LRRK2, GBA and their interaction in the regulation of autophagy: Implications on therapeutics in Parkinson’s disease. Transl. Neurodegener. 2022, 11, 5. [Google Scholar] [CrossRef]
- Azeggagh, S.; Berwick, D.C. The development of inhibitors of leucine-rich repeat kinase 2 (LRRK2) as a therapeutic strategy for Parkinson’s disease: The current state of play. Br. J. Pharmacol. 2022, 179, 1478–1495. [Google Scholar] [CrossRef]
- Tang, X.; Xing, S.; Ma, M.; Xu, Z.; Guan, Q.; Chen, Y.; Feng, F.; Liu, W.; Chen, T.; Sun, H. The Development and Design Strategy of Leucine-Rich Repeat Kinase 2 Inhibitors: Promising Therapeutic Agents for Parkinson’s Disease. J. Med. Chem. 2023, 66, 2282–2307. [Google Scholar] [CrossRef]
- Gegg, M.E.; Menozzi, E.; Schapira, A.H.V. Glucocerebrosidase-associated Parkinson disease: Pathogenic mechanisms and potential drug treatments. Neurobiol. Dis. 2022, 166, 105663. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Submitted Protein * | Submitted Ligand ** | Vina Score *** | Cavity Volume (Å3) | Center (x, y, z) | Docking Size (x, y, z) | Contact Residue |
|---|---|---|---|---|---|---|
| GCase (6T13) | CBE (conformer 3D CID 9989541) | −6.0 | 3950 | 13, 10, −2 | 16, 34, 33 | Chain A: Asp127 Phe128 Trp179 Asn234 Glu235 Tyr244 Phe246 Gln284 Tyr313 Glu340 Cys342 Ser345 Trp381 Asn396 Val398 |
| ROT (conformer 3D CID 6758) | −9.2 | 3950 | 13, 10, −2 | 22, 34, 33 | Chain A: Asp127 Phe128 Trp179 Asn234 Glu235 Ser237 Ala238 Leu241 Tyr244 Pro245 Phe246 Asp283 Gln284 Tyr313 Leu314 Glu340 Cys342 Ser345 Trp381 Asn396 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Abshana, L.P.; Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Rotenone Blocks the Glucocerebrosidase Enzyme and Induces the Accumulation of Lysosomes and Autophagolysosomes Independently of LRRK2 Kinase in HEK-293 Cells. Int. J. Mol. Sci. 2023, 24, 10589. https://doi.org/10.3390/ijms241310589
Perez-Abshana LP, Mendivil-Perez M, Velez-Pardo C, Jimenez-Del-Rio M. Rotenone Blocks the Glucocerebrosidase Enzyme and Induces the Accumulation of Lysosomes and Autophagolysosomes Independently of LRRK2 Kinase in HEK-293 Cells. International Journal of Molecular Sciences. 2023; 24(13):10589. https://doi.org/10.3390/ijms241310589
Chicago/Turabian StylePerez-Abshana, Laura Patricia, Miguel Mendivil-Perez, Carlos Velez-Pardo, and Marlene Jimenez-Del-Rio. 2023. "Rotenone Blocks the Glucocerebrosidase Enzyme and Induces the Accumulation of Lysosomes and Autophagolysosomes Independently of LRRK2 Kinase in HEK-293 Cells" International Journal of Molecular Sciences 24, no. 13: 10589. https://doi.org/10.3390/ijms241310589