
Immune Cell-Related Genes in Juvenile Idiopathic Arthritis Identified Using Transcriptomic and Single-Cell Sequencing Data


 and
and
Abstract
:1. Introduction
2. Results
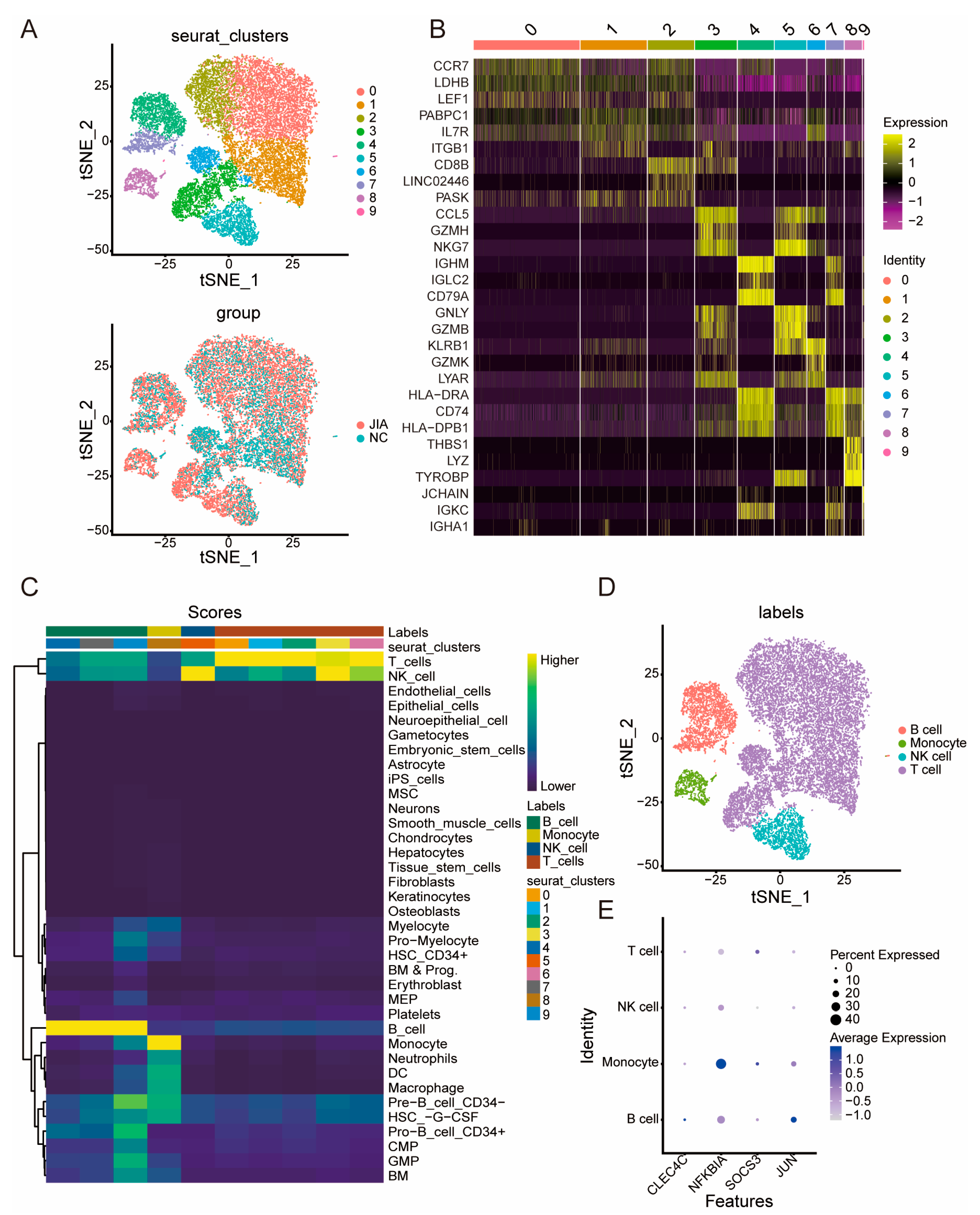
2.1. Immune Cells Are Significantly Different between NC and JIA Samples
2.2. A Total of 13,706 Genes Are Associated with JIA-Related Immune Cells
2.3. Differential Analysis Revealed 168 DE-ICRGs
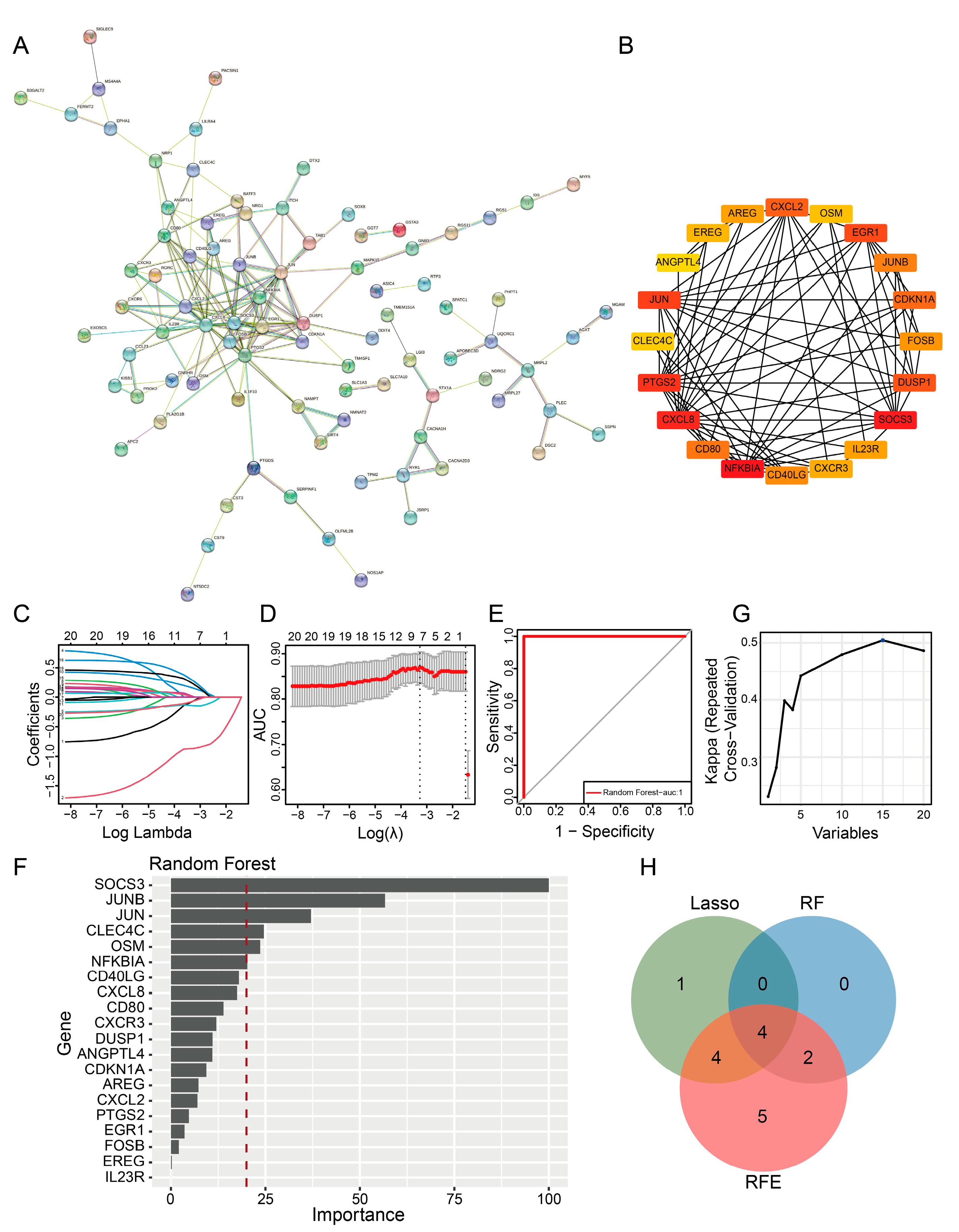
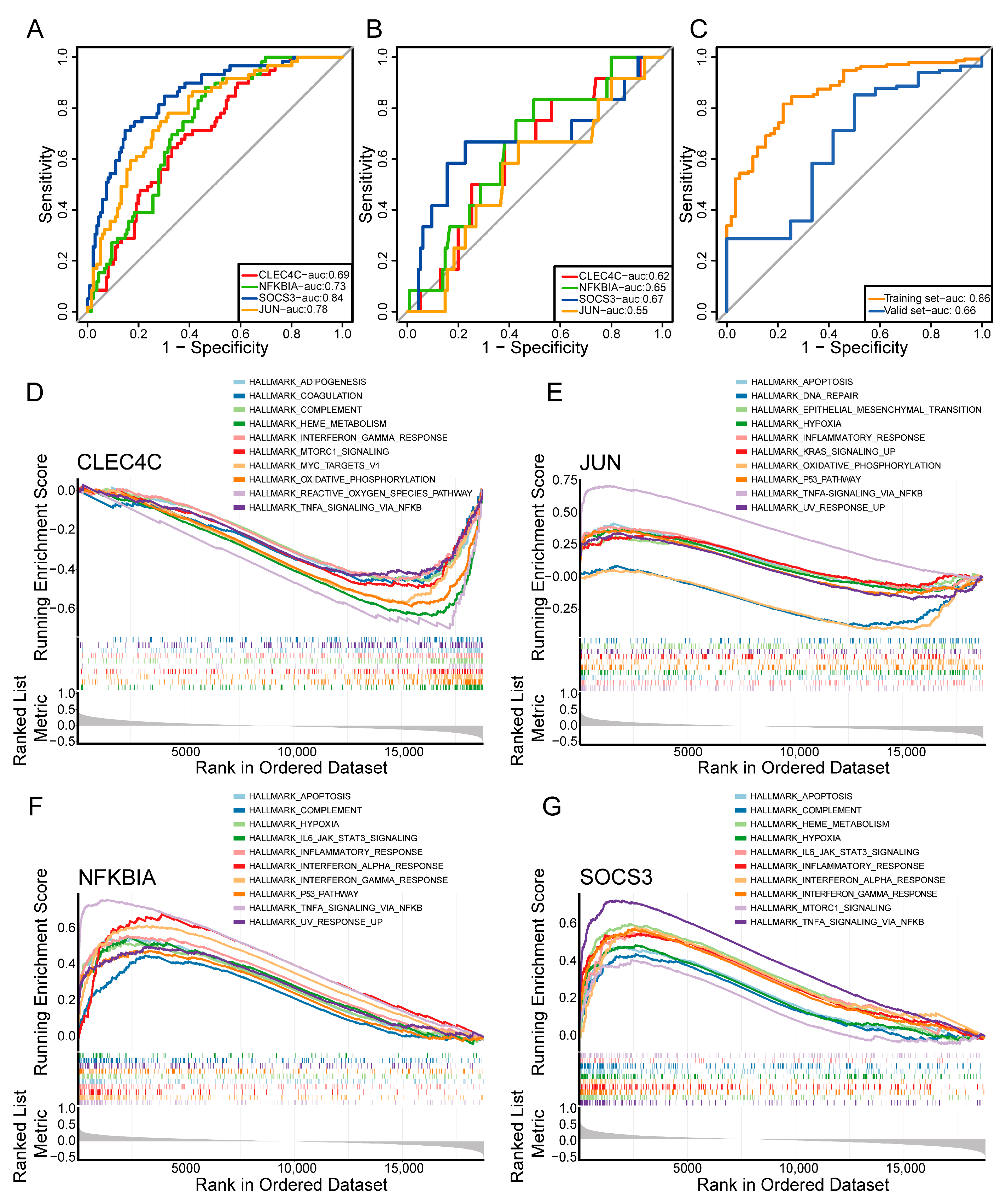
2.4. PPI Network and Machine Learning Analysis Revealed Key Prognostic Genes
2.5. ScRNA-Seq Analysis of NC and JIA Samples
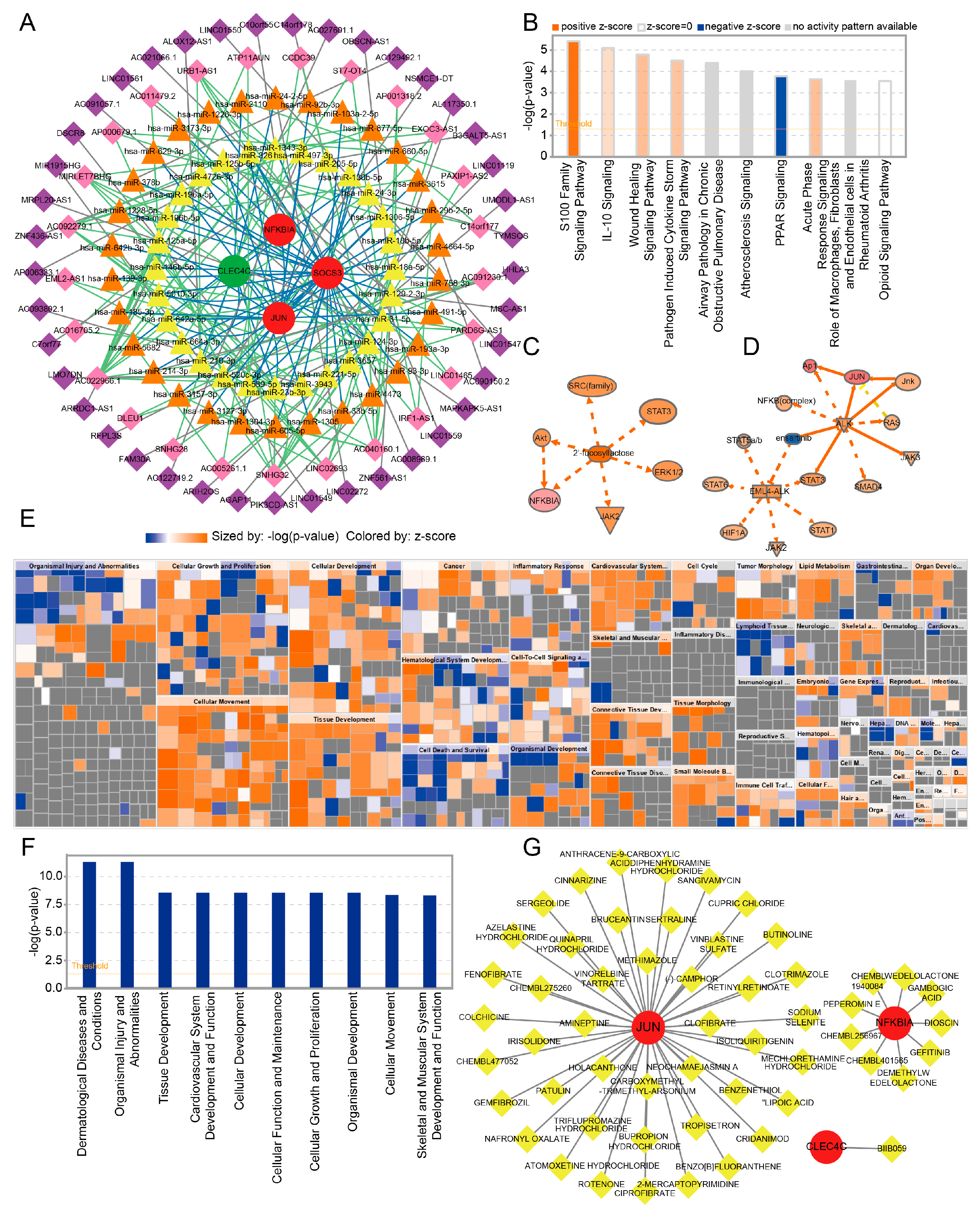
2.6. Construction of the ceRNA Network of Key Genes
2.7. IPA and Prediction of Potential Drugs Targeting the Key Genes
2.8. Validation of Key Genes by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Immune Microenvironment Analysis of NC and JIA Samples
4.3. Identification of Immune Cell-Related Genes (ICRGs)
4.4. Differential Expression and Enrichment Analysis of ICRGs
4.5. Protein–Protein Interaction (PPI) Network and Machine Learning Analysis of DE-ICRGs
4.6. ScRNA-Seq Analysis of NC and JIA Samples
4.7. Construction of a Competing Endogenous RNAs (ceRNA) Network
4.8. Ingenuity Pathway Analysis (IPA) and Prediction of Potential Drugs Targeting the Key Genes
4.9. In Vivo Experiments
4.10. Real-Time Quantitative PCR (RT-qPCR)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martini, A.; Lovell, D.J.; Albani, S.; Brunner, H.I.; Hyrich, K.L.; Thompson, S.D.; Ruperto, N. Juvenile idiopathic arthritis. Nat. Rev. Dis. Prim. 2022, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Consolaro, A.; Giancane, G.; Alongi, A.; van Dijkhuizen, E.H.P.; Aggarwal, A.; Al-Mayouf, S.M.; Bovis, F.; De Inocencio, J.; Demirkaya, E.; Flato, B.; et al. Phenotypic variability and disparities in treatment and outcomes of childhood arthritis throughout the world: An observational cohort study. Lancet Child Adolesc. Health 2019, 3, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Giancane, G.; Muratore, V.; Marzetti, V.; Quilis, N.; Benavente, B.S.; Bagnasco, F.; Alongi, A.; Civino, A.; Quartulli, L.; Consolaro, A.; et al. Disease activity and damage in juvenile idiopathic arthritis: Methotrexate era versus biologic era. Arthritis Res. Ther. 2019, 21, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Berthold, E.; Arve-Butler, S.; Gullstrand, B.; Mossberg, A.; Kahn, F.; Bengtsson, A.A.; Månsson, B.; Kahn, R. Children with oligoarticular juvenile idiopathic arthritis have skewed synovial monocyte polarization pattern with functional impairment-a distinct inflammatory pattern for oligoarticular juvenile arthritis. Arthritis Res. Ther. 2020, 22, 186. [Google Scholar] [CrossRef]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Zeng, L.; Yang, K.; Zhang, T.; Zhu, X.; Hao, W.; Chen, H.; Ge, J. Research progress of single-cell transcriptome sequencing in autoimmune diseases and autoinflammatory disease: A review. J. Autoimmun. 2022, 133, 102919. [Google Scholar] [CrossRef]
- Zaripova, L.N.; Midgley, A.; Christmas, S.E.; Beresford, M.W.; Baildam, E.M.; Oldershaw, R.A. Juvenile idiopathic arthritis: From aetiopathogenesis to therapeutic approaches. Pediatr. Rheumatol. Online J. 2021, 19, 135. [Google Scholar] [CrossRef]
- Dumaine, C.; Bekkar, S.; Belot, A.; Cabrera, N.; Malik, S.; von-Scheven, A.; Carbasse, A.; Woerner, A.; Wouters, C.; Bouayed, K.; et al. Infectious adverse events in children with Juvenile Idiopathic Arthritis treated with Biological Agents in a real-life setting: Data from the JIRcohorte. Jt. Bone Spine 2020, 87, 49–55. [Google Scholar] [CrossRef]
- Chen, H.; Ye, F.; Guo, G. Revolutionizing immunology with single-cell RNA sequencing. Cell Mol Immunol. 2019, 16, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.; Xi, Y.; Upham, J.W. CLEC4C gene expression can be used to quantify circulating plasmacytoid dendritic cells. J. Immunol. Methods 2019, 464, 126–130. [Google Scholar] [CrossRef]
- Hoober, J.K.; van-Vliet, S.J.; Dudziak, D.; Eggink, L.L. Editorial: Sentinel CLECs at Immunological Decision Nodes. Front. Immunol. 2020, 11, 2066. [Google Scholar] [CrossRef]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Günther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef]
- Pierre, N.; Huynh-Thu, V.A.; Marichal, T.; Allez, M.; Bouhnik, Y.; Laharie, D.; Bourreille, A.; Colombel, J.F.; Meuwis, M.A.; Louis, E. Distinct blood protein profiles associated with the risk of short-term and mid/long-term clinical relapse in patients with Crohn’s disease stopping infliximab: When the remission state hides different types of residual disease activity. Gut 2023, 72, 443–450. [Google Scholar] [CrossRef]
- Papavassiliou, A.G.; Musti, A.M. The Multifaceted Output of c-Jun Biological Activity: Focus at the Junction of CD8 T Cell Activation and Exhaustion. Cells 2020, 9, 2470. [Google Scholar] [CrossRef]
- Mao, R.; Fan, Y.; Mou, Y.; Zhang, H.; Fu, S.; Yang, J. TAK1 lysine 158 is required for TGF-β-induced TRAF6-mediated Smad-independent IKK/NF-κB and JNK/AP-1 activation. Cell. Signal. 2011, 23, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.B.; Cowley, C.J.; Sajjath, S.M.; Barrows, D.; Yang, Y.; Carroll, T.S.; Fuchs, E. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell 2021, 28, 1758–1774.e1758. [Google Scholar] [CrossRef]
- Novoszel, P.; Holcmann, M.; Stulnig, G.; De-Sa-Fernandes, C.; Zyulina, V.; Borek, I.; Linder, M.; Bogusch, A.; Drobits, B.; Bauer, T.; et al. Psoriatic skin inflammation is promoted by c-Jun/AP-1-dependent CCL2 and IL-23 expression in dendritic cells. EMBO Mol. Med. 2021, 13, e12409. [Google Scholar] [CrossRef]
- Yang, J.; Huang, M.; Zhou, L.; He, X.; Jiang, X.; Zhang, Y.; Xu, G. Cereblon suppresses the lipopolysaccharide-induced inflammatory response by promoting the ubiquitination and degradation of c-Jun. J. Biol. Chem. 2018, 293, 10141–10157. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, P.; Rivara, S.; Liu, C.; Ricci, J.; Ren, X.; Hurley, J.H.; Ablasser, A. Clathrinid-associated AP-1 controls termination of STING signalling. Nature 2022, 610, 761–767. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.L.; Zou, Y.F.; Feng, X.L.; Shi, H.J.; Du, X.F.; Shao, M.H.; Gu, Y.; Zhou, Q. Association of the NFKBIA gene polymorphisms with susceptibility to autoimmune and inflammatory diseases: A meta-analysis. Inflamm. Res. 2011, 60, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Sogkas, G.; Adriawan, I.R.; Ringshausen, F.C.; Baumann, U.; Schröder, C.; Klemann, C.; von Hardenberg, S.; Schmidt, G.; Bernd, A.; Jablonka, A.; et al. A novel NFKBIA variant substituting serine 36 of IκBα causes immunodeficiency with warts, bronchiectasis and juvenile rheumatoid arthritis in the absence of ectodermal dysplasia. Clin. Immunol. 2020, 210, 108269. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Sun, Y.; Fu, X.; Wang, Z.; Yu, G.; Yue, Z.; Wang, Y.; Zhang, H.; Wang, C.; Liu, H.; et al. Identification of a Single Nucleotide Polymorphism in NFKBIA with Different Effects on Psoriatic Arthritis and Cutaneous Psoriasis in China. Acta Derm.-Venereol. 2019, 99, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.T.; Jung, I.D.; Cha, G.S.; Han, M.K.; Park, Y.M. Platelet-activating Factor Mediates Endotoxin Tolerance by Regulating Indoleamine 2,3-Dioxygenase-dependent Expression of the Suppressor of Cytokine Signaling 3. J. Biol. Chem. 2017, 292, 3290–3298. [Google Scholar] [CrossRef] [Green Version]
- Martino, N.; Ramos, R.B.; Lu, S.; Leyden, K.; Tomaszek, L.; Sadhu, S.; Fredman, G.; Jaitovich, A.; Vincent, P.A.; Adam, A.P. Endothelial SOCS3 maintains homeostasis and promotes survival in endotoxemic mice. J. Clin. Investig. 2021, 6, e147280. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, H.; Wang, P.; Wang, J.; Zou, L. The roles of SOCS3 and STAT3 in bacterial infection and inflammatory diseases. Scand. J. Immunol. 2018, 88, e12727. [Google Scholar] [CrossRef] [Green Version]
- Gattorno, M.; Chicha, L.; Gregorio, A.; Ferlito, F.; Rossi, F.; Jarrossay, D.; Lanzavecchia, A.; Martini, A.; Manz, M.G. Distinct expression pattern of IFN-alpha and TNF-alpha in juvenile idiopathic arthritis synovial tissue. Rheumatology 2007, 46, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, D.; Tenbrock, K.; Roth, J. Alarmins of the S100-Family in Juvenile Autoimmune and Auto-Inflammatory Diseases. Front. Immunol. 2019, 10, 182. [Google Scholar] [CrossRef]
- Tao, J.; Kamanaka, M.; Hao, J.; Hao, Z.; Jiang, X.; Craft, J.E.; Flavell, R.A.; Wu, Z.; Hong, Z.; Zhao, L.; et al. IL-10 signaling in CD4+ T cells is critical for the pathogenesis of collagen-induced arthritis. Arthritis Res. Ther. 2011, 13, R212. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, J.; Zhou, J.; Feng, J.; Zhuang, W.; Chen, J.; Zhao, J.; Zhong, W.; Zhao, Y.; Zhang, Y.; et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell lung cancer: A multicentre, phase 2 trial. Lancet Respir. Med. 2020, 8, 45–53. [Google Scholar] [CrossRef]
- Eiselein, L.; Nyunt, T.; Lamé, M.W.; Ng, K.F.; Wilson, D.W.; Rutledge, J.C.; Aung, H.H. TGRL Lipolysis Products Induce Stress Protein ATF3 via the TGF-β Receptor Pathway in Human Aortic Endothelial Cells. PLoS ONE 2015, 10, e0145523. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wan, L.; Li, W.; Ni, D.; Zhang, W.; Yan, X.; Mu, W. Recent advances on 2’-fucosyllactose: Physiological properties, applications, and production approaches. Crit. Rev. Food Sci. Nutr. 2022, 62, 2083–2092. [Google Scholar] [CrossRef]
- Yao, Q.; Fan, L.; Zheng, N.; Blecker, C.; Delcenserie, V.; Li, H.; Wang, J. 2′-Fucosyllactose Ameliorates Inflammatory Bowel Disease by Modulating Gut Microbiota and Promoting MUC2 Expression. Front. Nutr. 2022, 9, 822020. [Google Scholar] [CrossRef]
- Lei, K.; Wang, D.; Lin, L.; Zeng, J.; Li, Y.; Zhang, L.; Lane, J.A.; Zuo, D.; Sun, L. 2′-fucosyllactose inhibits imiquimod-induced psoriasis in mice by regulating Th17 cell response via the STAT3 signaling pathway. Int. Immunopharmacol. 2020, 85, 106659. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, D.; Guan, Y.; Wang, Z.; Sang, D.; Liu, M.; Pu, Y.; Fang, X.; Wang, D.; Huang, A.; et al. Disulfiram and Diphenhydramine Hydrochloride Upregulate miR-30a to Suppress IL-17-Associated Autoimmune Inflammation. J. Neurosci. 2016, 36, 9253–9266. [Google Scholar] [CrossRef] [Green Version]
- Karimollah, A.; Hemmatpur, A.; Hosseini, N.; Manshadi, M.D. Tropisetron balances immune responses via TLR2, TLR4 and JAK2/STAT3 signalling pathway in LPS-stimulated PBMCs. Basic Clin. Pharmacol. Toxicol. 2021, 128, 669–676. [Google Scholar] [CrossRef]
- Rada, J.; Donato, M.; Penas, F.N.; Alba-Soto, C.; Cevey, Á.C.; Pieralisi, A.V.; Gelpi, R.; Mirkin, G.A.; Goren, N.B. IL-10-Dependent and -Independent Mechanisms Are Involved in the Cardiac Pathology Modulation Mediated by Fenofibrate in an Experimental Model of Chagas Heart Disease. Front. Immunol. 2020, 11, 572178. [Google Scholar] [CrossRef]
- Zhou, Z.; Liang, Y.; Gao, Y.; Kong, W.; Feng, J.; Wang, X. Fenofibrate enhances the in vitro differentiation of foxp3(+) regulatory T cells in mice. PPAR Res. 2012, 2012, 529035. [Google Scholar] [PubMed] [Green Version]
- Ibarra-Lara, L.; Sánchez-Aguilar, M.; Soria-Castro, E.; Vargas-Barrón, J.; Roldán, F.J.; Pavón, N.; Torres-Narváez, J.C.; Cervantes-Pérez, L.G.; Pastelín-Hernández, G.; Sánchez-Mendoza, A. Clofibrate Treatment Decreases Inflammation and Reverses Myocardial Infarction-Induced Remodelation in a Rodent Experimental Model. Molecules 2019, 24, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xu, F.; Hu, J.; Zhang, H.; Cui, L.; Lu, W.; He, W.; Wang, X.; Li, M.; Zhang, H.; et al. Modulation of lactate-lysosome axis in dendritic cells by clotrimazole potentiates antitumor immunity. J. Immunother. Cancer 2021, 9, e002155. [Google Scholar] [CrossRef] [PubMed]
- Slobodnick, A.; Shah, B.; Krasnokutsky, S.; Pillinger, M.H. Update on colchicine, 2017. Rheumatology 2018, 57 (Suppl. 1), i4–i11. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Lin, Y.; Dou, J.; Fu, Z.; Yao, Y.; Ye, S.; Zhang, S.; Wang, N.; Liu, A.; Li, X.; et al. Wedelolactone facilitates Ser/Thr phosphorylation of NLRP3 dependent on PKA signalling to block inflammasome activation and pyroptosis. Cell Prolif. 2020, 53, e12868. [Google Scholar] [CrossRef]
- Yin, W.; Liu, S.; Dong, M.; Liu, Q.; Shi, C.; Bai, H.; Wang, Q.; Yang, X.; Niu, W.; Wang, L. A New NLRP3 Inflammasome Inhibitor, Dioscin, Promotes Osteogenesis. Small 2020, 16, e1905977. [Google Scholar] [CrossRef]
- Yan, J.; Li, M.; Wang, X.D.; Lu, Z.Y.; Ni, X.L. Peperomin E (PepE) protects against high fat diet-induced atherosclerosis in Apolipoprotein E deficient (ApoE(-/-)) mice through reducing inflammation via the suppression of NLRP3 signaling pathway. Biomed. Pharmacother. 2018, 105, 862–869. [Google Scholar] [CrossRef]
- Wang, Q.L.; Yang, D.Z.; Lv, C. Anti-inflammatory effects of gambogic acid in murine collagen-induced arthritis through PI3K/Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4791–4796. [Google Scholar] [CrossRef] [Green Version]
- Furie, R.; Werth, V.P.; Merola, J.F.; Stevenson, L.; Reynolds, T.L.; Naik, H.; Wang, W.; Christmann, R.; Gardet, A.; Pellerin, A.; et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Investig. 2019, 129, 1359–1371. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, M.; Kim, H.; Lee, Y.; Lee, Y.I. Phytochemical and Pharmacological Role of Liquiritigenin and Isoliquiritigenin From Radix Glycyrrhizae in Human Health and Disease Models. Front. Aging. Neurosci. 2018, 10, 348. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.M.; Park, K.T.; Noh, H.D.; Lee, S.H.; Kim, D.H. Kakkalide and irisolidone alleviate 2,4,6-trinitrobenzenesulfonic acid-induced colitis in mice by inhibiting lipopolysaccharide binding to toll-like receptor-4 and proteobacteria population. Int. Immunopharmacol. 2019, 73, 246–253. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhang, H.; Tang, X.; Feng, R.; Yao, G.; Chen, W.; Li, W.; Liang, J.; Feng, X.; Sun, L. Mesenchymal stem cells promote the osteogenesis in collagen-induced arthritic mice through the inhibition of TNF-α. Stem Cells Int. 2018, 2018, 4069032. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Murphy, D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e79. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence |
|---|---|
| Nfkbia F | ATGGAAGTCATTGGTCAGGTGA |
| Nfkbia R | ACAGGCAAGATGTAGAGGGGTA |
| Socs3 F | CAGTCGGGGACCAAGAACCTA |
| Socs3 R | GTACACAGTCGAAGCGGGGAA |
| Jun F | ACGCCAACCTCAGCAACTT |
| Jun R | TCCTCATGCGCTTCCTCTC |
| Internal reference-M-Gapdh F | CCTTCCGTGTTCCTACCCC |
| Internal reference-M-Gapdh R | GCCCAAGATGCCCTTCAGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Cai, Z.; Liang, D.; Han, J.; Wu, P.; Shan, J.; Meng, G.; Zeng, H. Immune Cell-Related Genes in Juvenile Idiopathic Arthritis Identified Using Transcriptomic and Single-Cell Sequencing Data. Int. J. Mol. Sci. 2023, 24, 10619. https://doi.org/10.3390/ijms241310619
Zhang W, Cai Z, Liang D, Han J, Wu P, Shan J, Meng G, Zeng H. Immune Cell-Related Genes in Juvenile Idiopathic Arthritis Identified Using Transcriptomic and Single-Cell Sequencing Data. International Journal of Molecular Sciences. 2023; 24(13):10619. https://doi.org/10.3390/ijms241310619
Chicago/Turabian StyleZhang, Wenbo, Zhe Cai, Dandan Liang, Jiaochan Han, Ping Wu, Jiayi Shan, Guangxun Meng, and Huasong Zeng. 2023. "Immune Cell-Related Genes in Juvenile Idiopathic Arthritis Identified Using Transcriptomic and Single-Cell Sequencing Data" International Journal of Molecular Sciences 24, no. 13: 10619. https://doi.org/10.3390/ijms241310619
APA StyleZhang, W., Cai, Z., Liang, D., Han, J., Wu, P., Shan, J., Meng, G., & Zeng, H. (2023). Immune Cell-Related Genes in Juvenile Idiopathic Arthritis Identified Using Transcriptomic and Single-Cell Sequencing Data. International Journal of Molecular Sciences, 24(13), 10619. https://doi.org/10.3390/ijms241310619