

Extensive Bioinformatics Analyses Reveal a Phylogenetically Conserved Winged Helix (WH) Domain (Zτ) of Topoisomerase IIα, Elucidating Its Very High Affinity for Left-Handed Z-DNA and Suggesting Novel Putative Functions

 ,
, 

 ,
,
Abstract
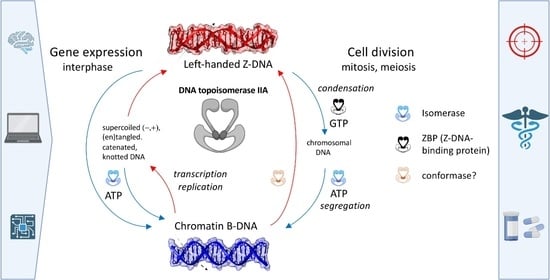
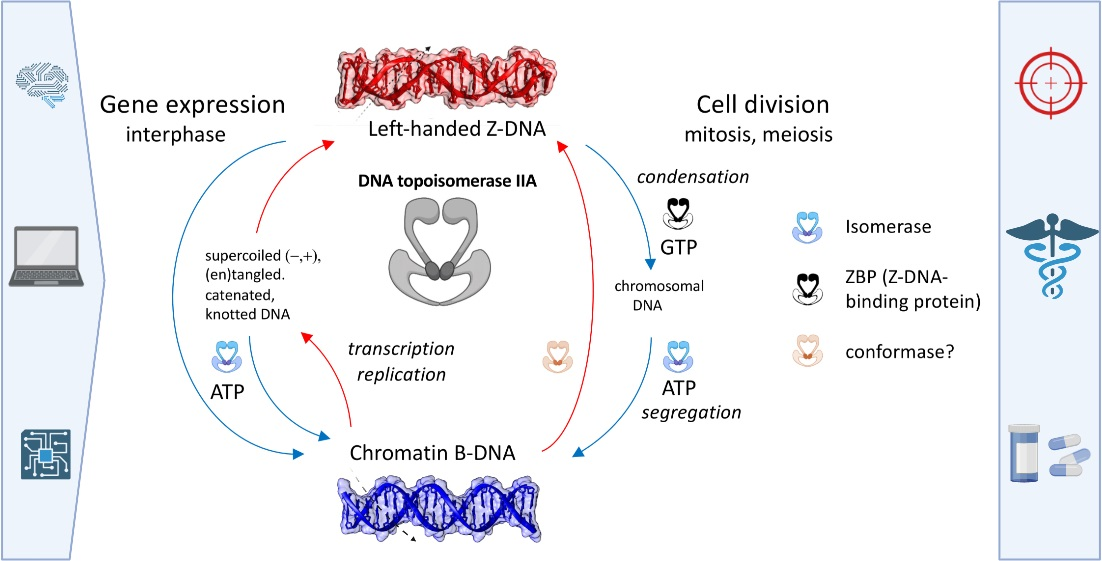
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Properties (1991–1994) a | Ref. |
|---|---|---|
| Drosophila (D) topoisomerase II and human (H) topoisomerase IIα (topoII) | ||
| 1 | Two orders of magnitude higher binding affinity for Z-DNA than for B-DNA (D); | [29] |
| 2 | Complexes with Z-DNA salt resistant after 5 min; | [29] |
| 3 | Inhibition by linear Z-DNA of relaxation of co-incubated nsc minicircles (D); | [33] |
| 4 | Preferential affinity for and enhanced relaxation of ns (D) minicircles with Z-DNA forming insert (D, H); | [33] |
| 5 | Distinct DNA loci of binding and scission (cleavage/resealing); | [33] |
| 6 | VM-26 inhibitor-induced covalent DNA–protein complexes with minicircles ± Z-DNA forming insert (D); | [33] |
| 7 | Much greater affinity for intrinsically curved compared to linear B-DNA (D, H); | [34] |
| 8 | Hierarchy of DNA affinity: linear Z-DNA ≈ curved B-DNA ≥ nscDNA >> linear B-DNA (D, H); | [34] |
| 9 | No binding of ssDNA (D); | [29] |
| 10 | Increased formation of aggregates of nsc minicircles with Z-DNA forming inserts (D). | [33] |
| Effects of GTP or non-hydrolyzable GTPγS (much more effective) | ||
| 11 | Persistent, time-dependent, temperature-dependent inactivation of enzyme activity (D); incubation ± nscDNA; | [29] |
| 12 | Inhibition of DNA relaxation activity (D, H, calf thymus) via a proposed allosteric mechanism; | [29] |
| 13 | A 5–10 increase in affinity for Z-DNA and decreased affinity for B-DNA (D); | [29] |
| 14 | Inhibition of ATPase activity (D); | [34] |
| 15 | Relaxation inhibited by >4 mM ATP and >0.5 mM ITP but not by UTP or CTP; | [29] |
| 16 | Limited DNA compaction (knotting, catenation) by stoichiometric Bombyx and human topoII; GTPase activity b. | [35] |
| human isoform topoisomerase IIβ | ||
| 17 | Hierarchy of DNA affinity: linear Z-DNA > nscDNA ≥ curved DNA >> poly[d(A-T)] > poly[d(G-C)]. | [34] |
2. Results and Discussion
2.1. TopoII Contains a Putative Z-DNA-Binding Domain (Zτ)

2.2. TopoII Contains a Major GTP-Binding Site
2.3. Both Zτ and GTP-Binding Site Are Phylogenetically Conserved across the Tree of Life


2.4. Molecular Docking of Various DNA Types to Zτ


| Zτ Docking Model | Zα Docking Model | Crystal | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | B-DNA | Z-DNA | B/Z-DNA | Z-RNA | B-DNA | Z-DNA | B/Z-DNA | Z-RNA | 4fm9 | 3f21 | |
| docking score 1 | −172 | −175 | −213 | −208 | −136 | −179 | −165 | −193 | - | - | |
| confidence score 2 | 0.61 | 0.62 | 0.78 | 0.76 | 0.43 | 0.64 | 0.57 | 0.70 | - | - | |
| no. contacts | 105 | 69 | 91 | 74 | 51 | 63 | 67 | 65 | 39 | 53 | |
| aa residue 3 | |||||||||||
| L722 n | 1 | Y136 a | 1 | 2 | 1 | K723 | K169 | ||||
| K723 b | 1, 2 | 1, 2 | 1 | 1, 2 | H159 b | 2 | Y757 | K170 | |||
| Q726 p | 2 | 1 | 2 | 2 | K169 b | 2 | 1, 2 | H759 | N173 | ||
| Y757 a | 2 | K170 b | 1 | 1 | 2 | S763 | R174 | ||||
| H759 b | 2 | 2 | 1 | 1 | N173 p | 1 | 1 | 2 | N770 | Y177 | |
| G760 n | 1 | 1 | R174 b | 1 | 1, 2 | 1 | 2 | K798 | T191 | ||
| M762 n | 1 | Y177 a | 2 | 1, 2 | 1 | 1, 2 | P192 | ||||
| S763 p | 2 | 2 | 1 | 1 | S178 p | 1 | 2 | 1 | P193 | ||
| T767 p | 2 | K181 b | 1 | 2 | 1 | ||||||
| N770 p | 2 | 1 | 1 | 2 | K187 b | 2 | 1 | 2 | |||
| L771 n | 2 | G190 n | 2 | ||||||||
| F775 n | 1 | T191 p | 1 | 1 2 | |||||||
| V776 n | 2 | 1 | 1 | P192 n | 2 | ||||||
| G777 n | 1 | 1, 2 | 1, 2 | P193 n | 1 | ||||||
| S778 p | 2 | 2 | |||||||||
| N779 p | 2 | 2 | |||||||||
| N780 p | 1, 2 | 1, 2 | 1, 2 | ||||||||
| L781 n | 2 | ||||||||||
| R793 b | 1 | ||||||||||
| G796 n | 1 | ||||||||||
| K798 b | 1 | 1 | 2 | ||||||||
| S802 p | 1 | ||||||||||
| R804 b | 1 | ||||||||||
| strand 1/(1 + 2) | 6/14 | 6/12 | 10/17 | 7/14 | 4/7 | 6/13 | 7/9 | 4/10 | 1 | 1 | |
2.5. Expanded “B-Z TopoII” Reaction Mechanism

2.6. A Case Study of the “B-Z TopoII” Mechanism: Mitosis
- What are the ultrastable topoII-DNA complexes that play a structural role in chromosome architecture? [43]
- Do centromeres drive chromosome compaction?
- How do non-B-DNA centromere sequences participate to (de)condensation?
- How does topoII contribute to axial shortening of the chromosomes [117]?
- How is cohesin release coordinated spatiotemporally with the actions of condensin and topoII in sister chromatid resolution [119]?
- How is large-scale compaction and spatial arrangement achieved [109]?
2.7. Perspectives and Biomedical Outlook
3. Materials and Methods
3.1. Structural Similarity Analysis of Human Z𝜏 (TOP2A) and Various Proteins Containing Zα Domains
3.2. Searching for Putative GTP-Binding Sites within Topoisomerases
3.3. Searching for Deleterious SNPs within Human TOP2A
3.4. Multiple Sequence Alignment of Full-Length TOP2 Protein Sequences
3.5. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, J.C. Untangling the Double Helix: DNA Entanglement and the Action of the DNA Topoisomerases; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; ISBN 978-0-87969-863-8. [Google Scholar]
- Hanke, A.; Ziraldo, R.; Levene, S.D. DNA-Topology Simplification by Topoisomerases. Molecules 2021, 26, 3375. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human Topoisomerases and Their Roles in Genome Stability and Organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Vidmar, V.; Vayssières, M.; Lamour, V. What’s on the Other Side of the Gate: A Structural Perspective on DNA Gate Opening of Type IA and IIA DNA Topoisomerases. Int. J. Mol. Sci. 2023, 24, 3986. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Sanderson, M. Principles of Nucleic Acid Structure, 2nd ed.; Academic Press: Cambridge, MA, USA, 2021; ISBN 978-0-12-819678-6. [Google Scholar]
- Du, Y.; Zhou, X. Targeting Non-B-Form DNA in Living Cells. Chem. Rec. 2013, 13, 371–384. [Google Scholar] [CrossRef]
- Pohl, F.M.; Jovin, T.M. Salt-Induced Co-Operative Conformational Change of a Synthetic DNA: Equilibrium and Kinetic Studies with Poly(dG-dC). J. Mol. Biol. 1972, 67, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Jovin, T.M. The Origin of Left-Handed Poly[d(G-C)]. In Z-DNA: Methods and Protocols; KIm, K.K., Subramani, V.K., Eds.; Springer: New York, NY, USA, 2023; pp. 1–32. [Google Scholar] [CrossRef]
- Wang, A.H.-J.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; van Boom, J.H.; van der Marel, G.; Rich, A. Molecular Structure of a Left-Handed Double Helical DNA Fragment at Atomic Resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Drew, H.; Takano, T.; Tanaka, S.; Itakura, K.; Dickerson, R.E. High-Salt d(CpGpCpG), a Left-Handed Z′ DNA Double Helix. Nature 1980, 286, 567–573. [Google Scholar] [CrossRef]
- Kim, D.; Subramani, V.K.; Park, S.; Lee, J.-H.; Kim, K.K. Z-DNA. In Handbook of Chemical Biology of Nucleic Acids; Sugimoto, N., Ed.; Springer Nature: Singapore, 2022; pp. 1–29. ISBN 9789811613135. [Google Scholar]
- Krall, J.B.; Nichols, P.J.; Henen, M.A.; Vicens, Q.; Vögeli, B. Structure and Formation of Z-DNA and Z-RNA. Molecules 2023, 28, 843. [Google Scholar] [CrossRef]
- KIm, K.K.; Subramani, V.K. (Eds.) Z-DNA: Methods and Protocols; Springer: New York, NY, USA, 2023. [Google Scholar] [CrossRef]
- Herbert, A. Z-DNA and Z-RNA in Human Disease. Commun. Biol. 2019, 2, 7. [Google Scholar] [CrossRef]
- Ravichandran, S.; Subramani, V.K.; Kim, K.K. Z-DNA in the Genome: From Structure to Disease. Biophys. Rev. 2019, 11, 383–387. [Google Scholar] [CrossRef]
- Kim, C. How Z-DNA/RNA Binding Proteins Shape Homeostasis, Inflammation, and Immunity. BMB Rep. 2020, 53, 453–457. [Google Scholar] [CrossRef]
- Herbert, A. The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity. Molecules 2021, 26, 4881. [Google Scholar] [CrossRef]
- Herbert, A. Z-DNA and Z-RNA: Methods—Past and Future. In Z-DNA: Methods and Protocols; Springer: New York, NY, USA, 2023; pp. 295–329. [Google Scholar] [CrossRef]
- Nichols, P.J.; Krall, J.B.; Henen, M.A.; Vögeli, B.; Vicens, Q. Z-RNA Biology: A Central Role in the Innate Immune Response? RNA 2023, 29, 273–281. [Google Scholar] [CrossRef]
- Herbert, A.; Alfken, J.; Kim, Y.-G.; Mian, I.S.; Nishikura, K.; Rich, A. A Z-DNA Binding Domain Present in the Human Editing Enzyme, Double-Stranded RNA Adenosine Deaminase. Proc. Natl. Acad. Sci. USA 1997, 94, 8421–8426. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.; Schade, M.; Lowenhaupt, K.; Alfken, J.; Schwartz, T.; Shlyakhtenko, L.S.; Lyubchenko, Y.L.; Rich, A. The Zα Domain from Human ADAR1 Binds to the Z-DNA Conformer of Many Different Sequences. Nucleic Acids Res. 1998, 26, 3486–3493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C. ADAR1 Masks the Cancer Immunotherapeutic Promise of ZBP1-Driven Necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.; Rould, M.A.; Lowenhaupt, K.; Herbert, A.; Rich, A. Crystal Structure of the Zα Domain of the Human Editing Enzyme ADAR1 Bound to Left-Handed Z-DNA. Science 1999, 284, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.C.; Li, Y.; Ng, S.K. The Role of the Z-DNA Binding Domain in Innate Immunity and Stress Granules. Front. Immunol. 2021, 11, 3779. [Google Scholar] [CrossRef]
- Meng, Y.; Wang, G.; He, H.; Lau, K.H.; Hurt, A.; Bixler, B.J.; Parham, A.; Jin, S.-G.; Xu, X.; Vasquez, K.M. Z-DNA Is Remodelled by ZBTB43 in Prospermatogonia to Safeguard the Germline Genome and Epigenome. Nat. Cell Biol. 2022, 24, 1141–1153. [Google Scholar] [CrossRef]
- Bartas, M.; Slychko, K.; Brázda, V.; Červeň, J.; Beaudoin, C.A.; Blundell, T.L.; Pečinka, P. Searching for New Z-DNA/Z-RNA Binding Proteins Based on Structural Similarity to Experimentally Validated Zα Domain. Int. J. Mol. Sci. 2022, 23, 768. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, R.; Kinebuchi, T.; Sato, M.; Yagi, H.; Kurumizaka, H.; Yokoyama, S. Stimulation of DNA Strand Exchange by the Human TBPIP/Hop2-Mnd1 Complex. J. Biol. Chem. 2006, 281, 5575–5581. [Google Scholar] [CrossRef] [PubMed]
- EMBL SMART Z-Alpha. Available online: http://smart.embl-heidelberg.de/smart/selective.cgi?domains=Zalpha&terms=&taxon_text=&input=Architecture+query (accessed on 10 February 2023).
- Arndt-Jovin, D.J.; Udvardy, A.; Garner, M.M.; Ritter, S.; Jovin, T.M. Z-DNA Binding and Inhibition by GTP of Drosophila Topoisomerase II. Biochemistry 1993, 32, 4862–4872. [Google Scholar] [CrossRef]
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA Topoisomerases: Advances in Understanding of Cellular Roles and Multi-Protein Complexes via Structure-Function Analysis. BioEssays 2021, 43, 2000286. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Lee, M.P.; Hsieh, T.; Griffith, J.D. Drosophila Topoisomerase II-DNA Interactions Are Affected by DNA Structure. J. Mol. Biol. 1991, 217, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Bigman, L.S.; Greenblatt, H.M.; Levy, Y. What Are the Molecular Requirements for Protein Sliding along DNA? J. Phys. Chem. B 2021, 125, 3119–3131. [Google Scholar] [CrossRef] [PubMed]
- Glikin, G.C.; Jovin, T.M.; Arndt-Jovin, D.J. Interactions of Drosophilla DNA Topoisomerase II with Left-Handed Z-DNA in Supercoiled Minicircles. Nucleic Acids Res. 1991, 19, 7139–7144. [Google Scholar] [CrossRef]
- Bechert, T.; Diekmann, S.; Arndt-Jovin, D.J. Human 170 KDa and 180 KDa Topoisomerases II Bind Preferentially to Curved and Left-Handed Linear DNA. J. Biomol. Struct. 1994, 12, 605–623. [Google Scholar] [CrossRef]
- Hirose, S.; Tabuchi, H.; Yoshinaga, K. GTP Induces Knotting, Catenation, and Relaxation of DNA by Stoichiometric Amounts of DNA Topoisomerase II from Bombyx Mori and HeLa Cells. J. Biol. Chem. 1988, 263, 3805–3810. [Google Scholar] [CrossRef]
- Bollimpelli, V.S.; Dholaniya, P.S.; Kondapi, A.K. Topoisomerase IIβ and Its Role in Different Biological Contexts. Arch. Biochem. Biophys. 2017, 633, 78–84. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, M.A.; Osheroff, N. DNA Topology and Topoisomerases: Teaching a “Knotty” Subject. Biochem. Mol. Biol. Educ. 2009, 37, 2–10. [Google Scholar] [CrossRef]
- Seol, Y.; Neuman, K.C. The Dynamic Interplay between DNA Topoisomerases and DNA Topology. Biophys. Rev. 2016, 8, 101–111. [Google Scholar] [CrossRef]
- Michieletto, D.; Fosado, Y.A.G.; Melas, E.; Baiesi, M.; Tubiana, L.; Orlandini, E. Dynamic and Facilitated Binding of Topoisomerase Accelerates Topological Relaxation. Nucleic Acids Res. 2022, 50, 4659–4668. [Google Scholar] [CrossRef]
- Antoniou-Kourounioti, M.; Mimmack, M.L.; Porter, A.C.; Farr, C.J. The Impact of the C-Terminal Region on the Interaction of Topoisomerase II Alpha with Mitotic Chromatin. Int. J. Mol. Sci. 2019, 20, 1238. [Google Scholar] [CrossRef]
- Hoang, K.G.; Menzie, R.A.; Rhoades, J.H.; Fief, C.A.; Deweese, J.E. Reviewing the Modification, Interactions, and Regulation of the C-Terminal Domain of Topoisomerase IIα as a Prospect for Future Therapeutic Targeting. EC Pharmacol. Toxicol. 2020, 8, 27–43. [Google Scholar]
- Hirsch, J.; Klostermeier, D. What Makes a Type IIA Topoisomerase a Gyrase or a Topo IV? Nucleic Acids Res. 2021, 49, 6027–6042. [Google Scholar] [CrossRef] [PubMed]
- Roca, J. Topoisomerase II: A Fitted Mechanism for the Chromatin Landscape. Nucleic Acids Res. 2009, 37, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The Structure of DNA-Bound Human Topoisomerase II Alpha: Conformational Mechanisms for Coordinating Inter-Subunit Interactions with DNA Cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef]
- Chen, S.-F.; Huang, N.-L.; Lin, J.-H.; Wu, C.-C.; Wang, Y.-R.; Yu, Y.-J.; Gilson, M.K.; Chan, N.-L. Structural Insights into the Gating of DNA Passage by the Topoisomerase II DNA-Gate. Nat. Commun. 2018, 9, 3085. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Berger, J.M. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes 2019, 10, 859. [Google Scholar] [CrossRef]
- Ziraldo, R.; Hanke, A.; Levene, S.D. Kinetic Pathways of Topology Simplification by Type-II Topoisomerases in Knotted Supercoiled DNA. Nucleic Acids Res. 2019, 47, 69–84. [Google Scholar] [CrossRef]
- Vanden Broeck, A.; Lotz, C.; Drillien, R.; Haas, L.; Bedez, C.; Lamour, V. Structural Basis for Allosteric Regulation of Human Topoisomerase IIα. Nat. Commun. 2021, 12, 2962. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.H.; Osheroff, N.; Berger, J.M. Structure of a Topoisomerase II–DNA–Nucleotide Complex Reveals a New Control Mechanism for ATPase Activity. Nat. Struct. Mol. Biol. 2012, 19, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The DNA Cleavage Reaction of Topoisomerase II: Wolf in Sheep’s Clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jung, S.-R.; Heo, K.; Byl, J.A.W.; Deweese, J.E.; Osheroff, N.; Hohng, S. DNA Cleavage and Opening Reactions of Human Topoisomerase IIα Are Regulated via Mg2+-Mediated Dynamic Bending of Gate-DNA. Proc. Natl. Acad. Sci. USA 2012, 109, 2925–2930. [Google Scholar] [CrossRef]
- Singh, B.N.; Achary, V.M.M.; Venkatapuram, A.K.; Parmar, H.; Karippadakam, S.; Sopory, S.K.; Reddy, M.K. Expression and Functional Analysis of Various Structural Domains of Tobacco Topoisomerase II: To Understand the Mechanistic Insights of Plant Type II Topoisomerases. Plant Physiol. Biochem. 2023, 194, 302–314. [Google Scholar] [CrossRef]
- Le, T.T.; Wu, M.; Lee, J.H.; Bhatt, N.; Inman, J.T.; Berger, J.M.; Wang, M.D. Etoposide Promotes DNA Loop Trapping and Barrier Formation by Topoisomerase II. Nat. Chem. Biol. 2023, 19, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef]
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 2021, 60, 1630–1641. [Google Scholar] [CrossRef]
- Matias-Barrios, V.M.; Dong, X. The Implication of Topoisomerase II Inhibitors in Synthetic Lethality for Cancer Therapy. Pharmaceuticals 2023, 16, 94. [Google Scholar] [CrossRef]
- Okoro, C.O.; Fatoki, T.H. A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 2532. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Nitiss, J.L.; Pommier, Y. SUMO: A Swiss Army Knife for Eukaryotic Topoisomerases. Front. Mol. Biosci. 2022, 9, 871161. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jaroszewski, L.; Iyer, M.; Sedova, M.; Godzik, A. FATCAT 2.0: Towards a Better Understanding of the Structural Diversity of Proteins. Nucleic Acids Res. 2020, 48, W60–W64. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Mizianty, M.J.; Kurgan, L. Prediction and Analysis of Nucleotide-Binding Residues Using Sequence and Sequence-Derived Structural Descriptors. Bioinformatics 2012, 28, 331–341. [Google Scholar] [CrossRef]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-Dependent Domain Movement in the ATPase Domain of a Human Type IIA DNA Topoisomerase. J. Biol. Chem. 2005, 280, 37041–37047. [Google Scholar] [CrossRef]
- Stanger, F.V.; Dehio, C.; Schirmer, T. Structure of the N-Terminal Gyrase B Fragment in Complex with ADP⋅Pi Reveals Rigid-Body Motion Induced by ATP Hydrolysis. PLoS ONE 2014, 9, e107289. [Google Scholar] [CrossRef]
- Moreira, F.; Arenas, M.; Videira, A.; Pereira, F. Evolutionary History of TOPIIA Topoisomerases in Animals. J. Mol. Evol. 2022, 90, 149–165. [Google Scholar] [CrossRef]
- Valdés, A.; Coronel, L.; Martínez-García, B.; Segura, J.; Dyson, S.; Díaz-Ingelmo, O.; Micheletti, C.; Roca, J. Transcriptional Supercoiling Boosts Topoisomerase II-Mediated Knotting of Intracellular DNA. Nucleic Acids Res. 2019, 47, 6946–6955. [Google Scholar] [CrossRef]
- Lang, A.J.; Mirski, S.E.; Cummings, H.J.; Yu, Q.; Gerlach, J.H.; Cole, S.P. Structural Organization of the Human TOP2A and TOP2B Genes. Gene 1998, 221, 255–266. [Google Scholar] [CrossRef]
- Guglielmini, J.; Gaia, M.; Da Cunha, V.; Criscuolo, A.; Krupovic, M.; Forterre, P. Viral Origin of Eukaryotic Type IIA DNA Topoisomerases. Virus Evol. 2022, 8, veac097. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 Years of the SMART Protein Domain Annotation Resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Corpet, F. Multiple Sequence Alignment with Hierarchical Clustering. Nucleic Acids Res 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Zhao, R.; Shin, D.S.; Fiser, A.; Goldman, I.D. Identification of a Functionally Critical GXXG Motif and Its Relationship to the Folate Binding Site of the Proton-Coupled Folate Transporter (PCFT-SLC46A1). Am. J. Physiol. Cell Physiol. 2012, 303, C673–C681. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Clements, J.; Finn, R.D. Skylign: A Tool for Creating Informative, Interactive Logos Representing Sequence Alignments and Profile Hidden Markov Models. BMC Bioinform. 2014, 15, 7. [Google Scholar] [CrossRef]
- Afowowe, T.O.; Sakurai, Y.; Urata, S.; Zadeh, V.R.; Yasuda, J. Topoisomerase II as a Novel Antiviral Target against Panarenaviral Diseases. Viruses 2022, 15, 105. [Google Scholar] [CrossRef]
- Cui, W.; Braun, E.; Wang, W.; Tang, J.; Zheng, Y.; Slater, B.; Li, N.; Chen, C.; Liu, Q.; Wang, B.; et al. Structural Basis for GTP-Induced Dimerization and Antiviral Function of Guanylate-Binding Proteins. Proc. Natl. Acad. Sci. USA 2021, 118, e2022269118. [Google Scholar] [CrossRef]
- Chang, C.-C.; Wang, Y.-R.; Chen, S.-F.; Wu, C.-C.; Chan, N.-L. New Insights into DNA-Binding by Type IIA Topoisomerases. Curr. Opin. Struct. Biol. 2013, 23, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.N.P.; Iismaa, S.; Begg, G.; Freymann, D.M.; Graham, R.M.; Lorand, L. Conserved Tryptophan in the Core Domain of Transglutaminase Is Essential for Catalytic Activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2738–2742. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Cao, K.; Chen, Y.; Shen, H.; Liu, Z.; Qin, H.; Cai, J.; Gao, F.; Yang, Y. Nuclear Transglutaminase 2 Interacts with Topoisomerase II⍺ to Promote DNA Damage Repair in Lung Cancer Cells. J. Exp. Clin. Cancer Res. 2021, 40, 224. [Google Scholar] [CrossRef]
- Ogrizek, M.; Janežič, M.; Valjavec, K.; Perdih, A. Catalytic Mechanism of ATP Hydrolysis in the ATPase Domain of Human DNA Topoisomerase IIα. J. Chem. Inf. Model. 2022, 62, 3896–3909. [Google Scholar] [CrossRef] [PubMed]
- Jovin, T.M. Recognition Mechanisms of DNA-Specific Enzymes. Annu. Rev. Biochem. 1976, 45, 889–920. [Google Scholar] [CrossRef]
- Bae, S.; Kim, D.; Kim, K.K.; Kim, Y.-G.; Hohng, S. Intrinsic Z-DNA Is Stabilized by the Conformational Selection Mechanism of Z-DNA-Binding Proteins. J. Am. Chem. Soc. 2011, 133, 668–671. [Google Scholar] [CrossRef]
- Dong, K.C.; Berger, J.M. Structural Basis for Gate-DNA Recognition and Bending by Type IIA Topoisomerases. Nature 2007, 450, 1201–1205. [Google Scholar] [CrossRef]
- Lee, I.; Dong, K.C.; Berger, J.M. The Role of DNA Bending in Type IIA Topoisomerase Function. Nucleic Acids Res. 2013, 41, 5444–5456. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.H.; Santos, S.; Mitchenall, L.A.; Stuchinskaya, T.; Taylor, J.A.; Maxwell, A. DNA G-Segment Bending Is Not the Sole Determinant of Topology Simplification by Type II DNA Topoisomerases. Sci. Rep. 2014, 4, 6158. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, M.K.; Zvoda, V.; Holte, M.N.; Becker, N.A.; Peters, J.P.; Maher, L.J., III; Ansari, A. Evidence for a Bind-Then-Bend Mechanism for Architectural DNA Binding Protein YNhp6A. Nucleic Acids Res. 2019, 47, 2871–2883. [Google Scholar] [CrossRef]
- Dickerson, R.E. DNA Bending: The Prevalence of Kinkiness and the Virtues of Normality. Nucleic Acids Res. 1998, 26, 1906–1926. [Google Scholar] [CrossRef]
- Raskó, T.; Finta, C.; Kiss, A. DNA Bending Induced by DNA (Cytosine-5) Methyltransferases. Nucleic Acids Res. 2000, 28, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Harteis, S.; Schneider, S. Making the Bend: DNA Tertiary Structure and Protein-DNA Interactions. Int. J. Mol. Sci. 2014, 15, 12335–12363. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Yeou, S.; Lee, N.K. DNA Bending Force Facilitates Z-DNA Formation under Physiological Salt Conditions. J. Am. Chem. Soc. 2022, 144, 13137–13145. [Google Scholar] [CrossRef]
- Jang, Y.; Son, H.; Lee, S.-W.; Hwang, W.; Jung, S.-R.; Byl, J.A.W.; Osheroff, N.; Lee, S. Selection of DNA Cleavage Sites by Topoisomerase II Results from Enzyme-Induced Flexibility of DNA. Cell Chem. Biol. 2019, 26, 502–511.e3. [Google Scholar] [CrossRef]
- Ansevin, A.T.; Wang, A.H. Evidence for a New Z-Type Left-Handed DNA Helix: Properties of Z(WC)-DNA. Nucleic Acids Res. 1990, 18, 6119–6126. [Google Scholar] [CrossRef]
- Fuertes, M.A.; Cepeda, V.; Alonso, C.; Pérez, J.M. Molecular Mechanisms for the B−Z Transition in the Example of Poly[d(G−C)·d(G−C)] Polymers. A Critical Review. Chem. Rev. 2006, 106, 2045–2064. [Google Scholar] [CrossRef]
- Chakraborty, D.; Wales, D.J. Probing Helical Transitions in a DNA Duplex. Phys. Chem. Chem. Phys. 2017, 19, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Jovin, T.M.; McIntosh, L.P.; Arndt-Jovin, D.J.; Zarling, D.A.; Robert-Nicoud, M.; van de Sande, J.H.; Jorgenson, K.F.; Eckstein, F. Left-Handed DNA: From Synthetic Polymers to Chromosomes. J. Biomol. Struct. Dyn. 1983, 1, 21–57. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The Use of Divalent Metal Ions by Type II Topoisomerases. Metallomics 2010, 2, 450–459. [Google Scholar] [CrossRef]
- Fogg, J.M.; Randall, G.L.; Pettitt, B.M.; Sumners, D.W.L.; Harris, S.A.; Zechiedrich, L. Bullied No More: When and How DNA Shoves Proteins Around. Q. Rev. Biophys. 2012, 45, 257–299. [Google Scholar] [CrossRef]
- Vlahovicek, K.; Munteanu, M.G.; Pongor, S. Sequence-Dependent Modelling of Local DNA Bending Phenomena: Curvature Prediction and Vibrational Analysis. Genetica 1999, 106, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Hardin, A.H.; Sarkar, S.K.; Seol, Y.; Liou, G.F.; Osheroff, N.; Neuman, K.C. Direct Measurement of DNA Bending by Type IIA Topoisomerases: Implications for Non-Equilibrium Topology Simplification. Nucleic Acids Res. 2011, 39, 5729–5743. [Google Scholar] [CrossRef]
- Lee, M.; Kim, S.H.; Hong, S.-C. Minute Negative Superhelicity Is Sufficient to Induce the B-Z Transition in the Presence of Low Tension. Proc. Natl. Sci. Acad. USA 2010, 107, 4985–4990. [Google Scholar] [CrossRef]
- Fogg, J.M.; Catanese, D.J.; Randall, G.L.; Swick, M.C.; Zechiedrich, L. Differences Between Positively and Negatively Supercoiled DNA That Topoisomerases May Distinguish. In Mathematics of DNA Structure, Function and Interactions; Benham, C.J., Harvey, S., Olson, W.K., Sumners, D.W., Swigon, D., Eds.; The IMA Volumes in Mathematics and its Applications; Springer: New York, NY, USA, 2009; Volume 150, pp. 73–121. ISBN 978-1-4419-0669-4. [Google Scholar]
- Vologodskii, A. Theoretical Models of DNA Topology Simplification by Type IIA DNA Topoisomerases. Nucleic Acids Res. 2009, 37, 3125–3133. [Google Scholar] [CrossRef]
- Cofsky, J.C.; Soczek, K.M.; Knott, G.J.; Nogales, E.; Doudna, J.A. CRISPR-Cas9 Bends and Twists DNA to Read Its Sequence. Nat. Struct. Mol. Biol. 2022, 29, 395–402. [Google Scholar] [CrossRef]
- Stuchinskaya, T.; Mitchenall, L.A.; Schoeffler, A.J.; Corbett, K.D.; Berger, J.M.; Bates, A.D.; Maxwell, A. How Do Type II Topoisomerases Use ATP Hydrolysis to Simplify DNA Topology beyond Equilibrium? Investigating the Relaxation Reaction of Nonsupercoiling Type II Topoisomerases. J. Mol. Biol. 2009, 385, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Pohl, F.M. Hysteretic Behaviour of a Z-DNA-Antibody Complex. Biophys. Chem. 1987, 26, 385–390. [Google Scholar] [CrossRef]
- Igamberdiev, A.U.; Kleczkowski, L.A. Toward Understanding the Emergence of Life: A Dual Function of the System of Nucleotides in the Metabolically Closed Autopoietic Organization. Biosystems 2023, 224, 104837. [Google Scholar] [CrossRef]
- Padget, K.; Pearson, A.D.; Austin, C.A. Quantitation of DNA Topoisomerase IIalpha and Beta in Human Leukaemia Cells by Immunoblotting. Leukemia 2000, 14, 1997–2005. [Google Scholar] [CrossRef]
- Walther, N.; Hossain, M.J.; Politi, A.Z.; Koch, B.; Kueblbeck, M.; Ødegård-Fougner, Ø.; Lampe, M.; Ellenberg, J. A Quantitative Map of Human Condensins Provides New Insights into Mitotic Chromosome Architecture. J. Cell Biol. 2018, 217, 2309–2328. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.F.; Zhang, T.; Barisic, M.; Kalitsis, P.; Hudson, D.F. Topoisomerase IIα Is Essential for Maintenance of Mitotic Chromosome Structure. Proc. Natl. Acad. Sci. USA 2020, 117, 12131–12142. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A Pathway for Mitotic Chromosome Formation. Science 2018, 359, eaao6135. [Google Scholar] [CrossRef]
- Elbatsh, A.M.O.; Kim, E.; Eeftens, J.M.; Raaijmakers, J.A.; van der Weide, R.H.; García-Nieto, A.; Bravo, S.; Ganji, M.; Uit de Bos, J.; Teunissen, H.; et al. Distinct Roles for Condensin’s Two ATPase Sites in Chromosome Condensation. Mol. Cell 2019, 76, 724–737.e5. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Heald, R. Emergent Properties of Mitotic Chromosomes. Curr. Opin. Cell Biol. 2020, 64, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Man, T.; Witt, H.; Peterman, E.J.G.; Wuite, G.J.L. The Mechanics of Mitotic Chromosomes. Q. Rev. Biophys. 2021, 54, e10. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.R.; Hudson, D.F.; Cisneros-Soberanis, F.; Earnshaw, W.C. Mitotic Chromosomes. Semin. Cell Dev. Biol. 2021, 117, 7–29. [Google Scholar] [CrossRef]
- Dekker, B.; Dekker, J. Regulation of the Mitotic Chromosome Folding Machines. Biochemistry 2022, 479, 2153–2173. [Google Scholar] [CrossRef] [PubMed]
- Stray, J.E.; Lindsley, J.E. Biochemical Analysis of the Yeast Condensin Smc2/4 Complex: An ATPase That Promo.tes Knotting of Circular DNA. J. Biol. Chem. 2003, 278, 26238–26248. [Google Scholar] [CrossRef]
- Mora-Bermúdez, F.; Gerlich, D.; Ellenberg, J. Maximal Chromosome Compaction Occurs by Axial Shortening in Anaphase and Depends on Aurora Kinase. Nat. Cell Biol. 2007, 9, 822–831. [Google Scholar] [CrossRef]
- Baxter, J.; Aragón, L. A Model for Chromosome Condensation Based on the Interplay between Condensin and Topoisomerase II. Trends Genet. 2012, 28, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Roca, J.; Dyson, S.; Segura, J.; Valdés, A.; Martínez-García, B. Keeping Intracellular DNA Untangled: A New Role for Condensin? BioEssays 2022, 44, e2100187. [Google Scholar] [CrossRef] [PubMed]
- Samejima, K.; Samejima, I.; Vagnarelli, P.; Ogawa, H.; Vargiu, G.; Kelly, D.A.; de Lima Alves, F.; Kerr, A.; Green, L.C.; Hudson, D.F.; et al. Mitotic Chromosomes Are Compacted Laterally by KIF4 and Condensin and Axially by Topoisomerase IIα. J. Cell Biol. 2012, 199, 755–770. [Google Scholar] [CrossRef]
- Riccio, A.A.; Schellenberg, M.J.; Williams, R.S. Molecular Mechanisms of Topoisomerase 2 DNA–Protein Crosslink Resolution. Cell. Mol. Life Sci. 2020, 77, 81–91. [Google Scholar] [CrossRef]
- Shintomi, K.; Hirano, T. Sister Chromatid Resolution: A Cohesin Releasing Network and Beyond. Chromosoma 2010, 119, 459–467. [Google Scholar] [CrossRef]
- Le, T.T.; Wang, M.D. Topoisomerase II and Etoposide—A Tangled Tale. Nat. Chem. Biol. 2023, 19, 546–547. [Google Scholar] [CrossRef]
- Boulikas, T. Nature of DNA Sequences at the Attachment Regions of Genes to the Nuclear Matrix. J. Cell. Biochem. 1993, 52, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. Nucleosomes and Flipons Exchange Energy to Alter Chromatin Conformation, the Readout of Genomic Information, and Cell Fate. Bioessays 2022, 44, e2200166. [Google Scholar] [CrossRef] [PubMed]
- Wolff, D.W.; Bianchi-Smiraglia, A.; Nikiforov, M.A. Compartmentalization and Regulation of GTP in Control of Cellular Phenotypes. Trends Mol. Med. 2022, 28, 758–769. [Google Scholar] [CrossRef]
- Traut, T.W. Physiological Concentrations of Purines and Pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Kaláb, P.; Pralle, A.; Isacoff, E.Y.; Heald, R.; Weis, K. Analysis of a RanGTP-Regulated Gradient in Mitotic Somatic Cells. Nature 2006, 440, 697–701. [Google Scholar] [CrossRef]
- Kapoor, T.M. Metaphase Spindle Assembly. Biology 2017, 6, 8. [Google Scholar] [CrossRef]
- Ozugergin, I.; Piekny, A. Complementary Functions for the Ran Gradient during Division. Small GTPases 2021, 12, 177–187. [Google Scholar] [CrossRef]
- Spence, J.M.; Critcher, R.; Ebersole, T.A.; Valdivia, M.M.; Earnshaw, W.C.; Fukagawa, T.; Farr, C.J. Co-Localization of Centromere Activity, Proteins and Topoisomerase II within a Subdomain of the Major Human X Alpha-Satellite Array. EMBO J. 2002, 21, 5269–5280. [Google Scholar] [CrossRef]
- Kasinathan, S.; Henikoff, S. Non-B-Form DNA Is Enriched at Centromeres. Mol. Biol. Evol. 2018, 35, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Mellone, B.G.; Fachinetti, D. Diverse Mechanisms of Centromere Specification. Curr. Biol. 2021, 31, R1491–R1504. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yi, C.; Zhang, Z.; Su, H.; Liu, C.; Huang, Y.; Li, W.; Hu, X.; Liu, C.; Birchler, J.A.; et al. Non-B-Form DNA Tends to Form in Centromeric Regions and Has Undergone Changes in Polyploid Oat Subgenomes. Proc. Natl. Acad. Sci. USA 2023, 120, e2211683120. [Google Scholar] [CrossRef]
- Mills, W.E.; Spence, J.M.; Fukagawa, T.; Farr, C.J. Site-Specific Cleavage by Topoisomerase 2: A Mark of the Core Centromere. Int. J. Mol. Sci. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed]
- Biton, Y.Y. Effects of Protein-Induced Local Bending and Sequence Dependence on the Configurations of Supercoiled DNA Minicircles. J. Chem. Theory Comput. 2018, 14, 2063–2075. [Google Scholar] [CrossRef]
- Wang, L.H.-C.; Mayer, B.; Stemmann, O.; Nigg, E.A. Centromere DNA Decatenation Depends on Cohesin Removal and Is Required for Mammalian Cell Division. J. Cell Sci. 2010, 123, 806–813. [Google Scholar] [CrossRef]
- Chu, L.; Zhang, Z.; Mukhina, M.; Zickler, D.; Kleckner, N. Sister Chromatids Separate during Anaphase in a Three-Stage Program as Directed by Interaxis Bridges. Proc. Natl. Acad. Sci. USA 2022, 119, e2123363119. [Google Scholar] [CrossRef]
- Gibson, E.G.; Deweese, J.E. Structural and Biochemical Basis of Etoposide-Resistant Mutations in Topoisomerase IIα. Symmetry 2022, 14, 1309. [Google Scholar] [CrossRef]
- Jaramillo-Lambert, A.; Fabritius, A.S.; Hansen, T.J.; Smith, H.E.; Golden, A. The Identification of a Novel Mutant Allele of Topoisomerase II in Caenorhabditis Elegans Reveals a Unique Role in Chromosome Segregation during Spermatogenesis. Genetics 2016, 204, 1407–1422. [Google Scholar] [CrossRef]
- Zdraljevic, S.; Strand, C.; Seidel, H.S.; Cook, D.E.; Doench, J.G.; Andersen, E.C. Natural Variation in a Single Amino Acid Substitution Underlies Physiological Responses to Topoisomerase II Poisons. PLoS Genet. 2017, 13, e1006891. [Google Scholar] [CrossRef] [PubMed]
- Masullo, L.A.; Lopez, L.F.; Stefani, F.D. A Common Framework for Single-Molecule Localization Using Sequential Structured Illumination. Biophys. Rep. 2022, 2, 100036. [Google Scholar] [CrossRef]
- Austin, C.A.; Cowell, I.G.; Khazeem, M.M.; Lok, D.; Ng, H.T. TOP2B’s Contributions to Transcription. Biochem. Soc. Trans. 2021, 49, 2483–2493. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Bandak, A.; Lee, A.S.Y.; Austin, C.A.; Nitiss, J.L.; Berger, J.M. A Complex Suite of Loci and Elements in Eukaryotic Type II Topoisomerases Determine Selective Sensitivity to Distinct Poisoning Agents. Nucleic Acids Res. 2019, 47, 8163–8179. [Google Scholar] [CrossRef]
- Van Ravenstein, S.X.; Mehta, K.P.; Kavlashvili, T.; Byl, J.A.W.; Zhao, R.; Osheroff, N.; Cortez, D.; Dewar, J.M. Topoisomerase II Poisons Inhibit Vertebrate DNA Replication through Distinct Mechanisms. EMBO J. 2022, 41, e110632. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Schoepfer, J.; Radimerski, T.; Chène, P. Discovery of a New Class of Catalytic Topoisomerase II Inhibitors Targeting the ATP-Binding Site by Structure Based Design. Part I. Bioorg. Med. Chem. Lett. 2009, 19, 4014–4017. [Google Scholar] [CrossRef]
- Park, S.; Hwang, S.-Y.; Shin, J.; Jo, H.; Na, Y.; Kwon, Y. A Chromenone Analog as an ATP-Competitive, DNA Non-Intercalative Topoisomerase II Catalytic Inhibitor with Preferences toward the Alpha Isoform. ChemComm 2019, 55, 12857–12860. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.C.; Choi, J.; Hwang, H.-Y.; Rich, A.; Kim, Y.-G.; Kim, K.K. The Structures of Non-CG-Repeat Z-DNAs Co-Crystallized with the Z-DNA-Binding Domain, HZα ADAR1. Nucleic Acids Res. 2009, 37, 629–637. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for Integrated Sequence-Structure Analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl Variation Resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and Rendering Sequence and 3D Information from Atomic Structures of Proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein–Protein and Protein–DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Zheng, G.; Lu, X.-J.; Olson, W.K. Web 3DNA—A Web Server for the Analysis, Reconstruction, and Visualization of Three-Dimensional Nucleic-Acid Structures. Nucleic Acids Res. 2009, 37, W240–W246. [Google Scholar] [CrossRef]
- Patro, L.P.P.; Kumar, A.; Kolimi, N.; Rathinavelan, T. 3D-NuS: A Web Server for Automated Modeling and Visualization of Non-Canonical 3-Dimensional Nucleic Acid Structures. J. Mol. Biol. 2017, 429, 2438–2448. [Google Scholar] [CrossRef]
- Kim, D.; Hur, J.; Han, J.H.; Ha, S.C.; Shin, D.; Lee, S.; Park, S.; Sugiyama, H.; Kim, K.K. Sequence Preference and Structural Heterogeneity of BZ Junctions. Nucleic Acids Res. 2018, 46, 10504–10513. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]



| Feature | Parameter | Units | Domain | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Zα | Zβ | Zα1 | Zα2 | Zα | Zα | Zα | n.c. | n.c. | |||
| protein | ADAR1p150 | ZBP1 | PKZ | E3 | ORF112 | UFM1 | H4 | ||||
| RMSD | structure | Å | 2.4 | 2.7 | 2.7 | 3.0 | 2.6 | 2.6 | 2.7 | 3.2 | 2.8 |
| p-value | structure | 10−3 | 0.11 | 1.3 | 0.39 | 2.8 | 0.24 | 0.34 | 0.70 | 200 | 55 |
| equivalent positions | structure | no. | 64 | 62 | 60 | 61 | 60 | 61 | 60 | 39 | 50 |
| gaps | sequence | % | 30 | 32 | 34 | 33 | 34 | 32 | 34 | 39 | 7 |
| identity | sequence | % | 7.7 | 8.8 | 3.3 | 5.5 | 2.2 | 3.3 | 6.6 | 9.4 | 7.4 |
| similarity | sequence | % | 26 | 24 | 17 | 18 | 18 | 19 | 22 | 19 | 22 |
| Function | Activity | Target(s) |
|---|---|---|
| tf1 | isomerase: double-helix passage (ΔLk) | B-DNA |
| tf2 | high affinity recognition and stabilization of left-handed double helix; no covalent protein–DNA intermediate | Z-DNA, Z-RNA? |
| tf3 | conformase: induction of the right-to-left transition in double-helical sense | B-DNA, A-RNA? |
| tf4 | pronounced positive heterotropic allosteric role of GTP in tf2 and tf3 | topoII |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartas, M.; Slychko, K.; Červeň, J.; Pečinka, P.; Arndt-Jovin, D.J.; Jovin, T.M. Extensive Bioinformatics Analyses Reveal a Phylogenetically Conserved Winged Helix (WH) Domain (Zτ) of Topoisomerase IIα, Elucidating Its Very High Affinity for Left-Handed Z-DNA and Suggesting Novel Putative Functions. Int. J. Mol. Sci. 2023, 24, 10740. https://doi.org/10.3390/ijms241310740
Bartas M, Slychko K, Červeň J, Pečinka P, Arndt-Jovin DJ, Jovin TM. Extensive Bioinformatics Analyses Reveal a Phylogenetically Conserved Winged Helix (WH) Domain (Zτ) of Topoisomerase IIα, Elucidating Its Very High Affinity for Left-Handed Z-DNA and Suggesting Novel Putative Functions. International Journal of Molecular Sciences. 2023; 24(13):10740. https://doi.org/10.3390/ijms241310740
Chicago/Turabian StyleBartas, Martin, Kristyna Slychko, Jiří Červeň, Petr Pečinka, Donna J. Arndt-Jovin, and Thomas M. Jovin. 2023. "Extensive Bioinformatics Analyses Reveal a Phylogenetically Conserved Winged Helix (WH) Domain (Zτ) of Topoisomerase IIα, Elucidating Its Very High Affinity for Left-Handed Z-DNA and Suggesting Novel Putative Functions" International Journal of Molecular Sciences 24, no. 13: 10740. https://doi.org/10.3390/ijms241310740
APA StyleBartas, M., Slychko, K., Červeň, J., Pečinka, P., Arndt-Jovin, D. J., & Jovin, T. M. (2023). Extensive Bioinformatics Analyses Reveal a Phylogenetically Conserved Winged Helix (WH) Domain (Zτ) of Topoisomerase IIα, Elucidating Its Very High Affinity for Left-Handed Z-DNA and Suggesting Novel Putative Functions. International Journal of Molecular Sciences, 24(13), 10740. https://doi.org/10.3390/ijms241310740