
In Silico Screening and Anticancer-Apoptotic Evaluation of Newly Synthesized Thienopyrimidine/Sulfonamide Hybrids
 ,
,
Abstract
:1. Introduction
2. Results and Discussion
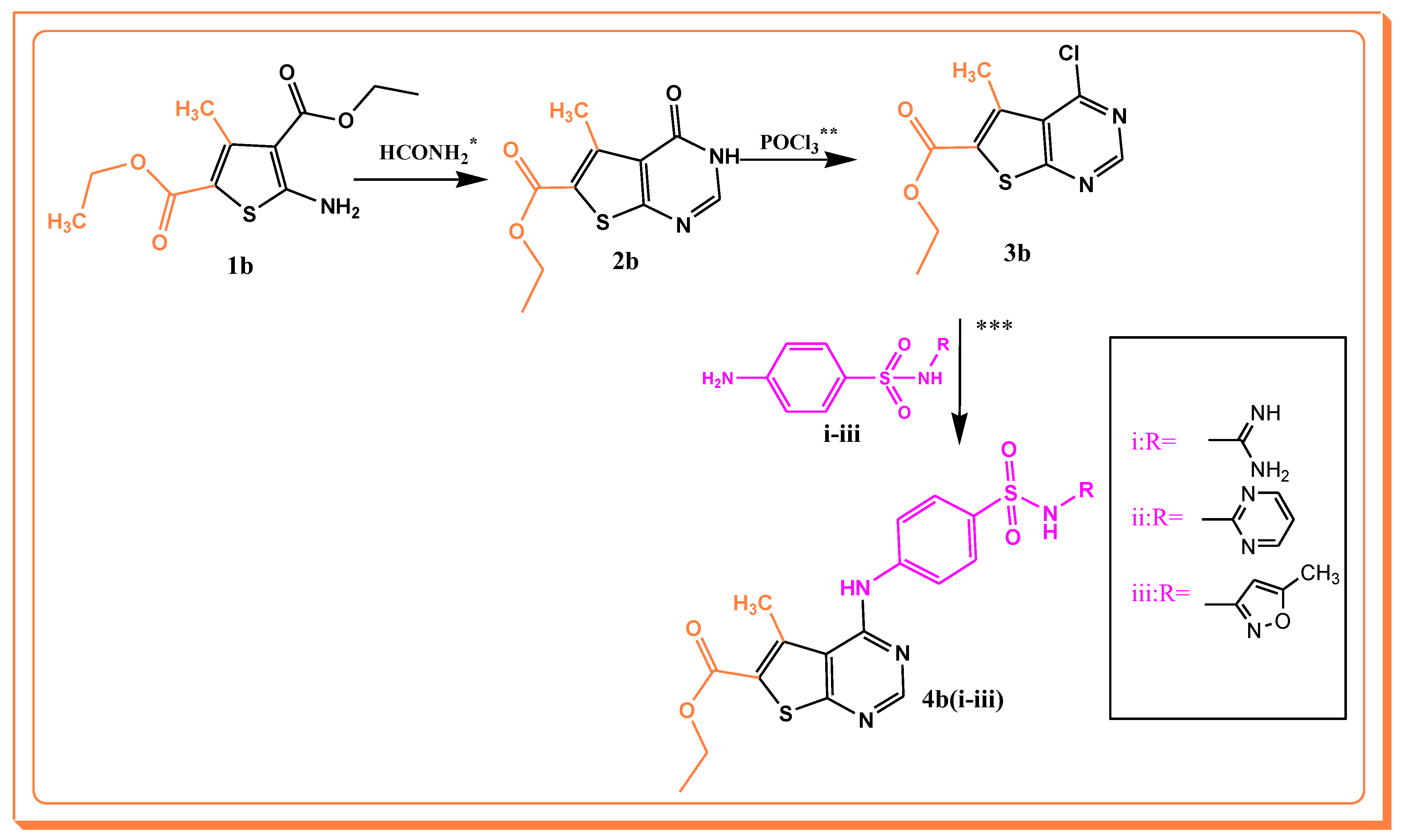
2.1. Synthesis
2.2. Biological Evaluation
2.2.1. In Vitro Anti-Proliferative Activity
2.2.2. Selectivity Assessment
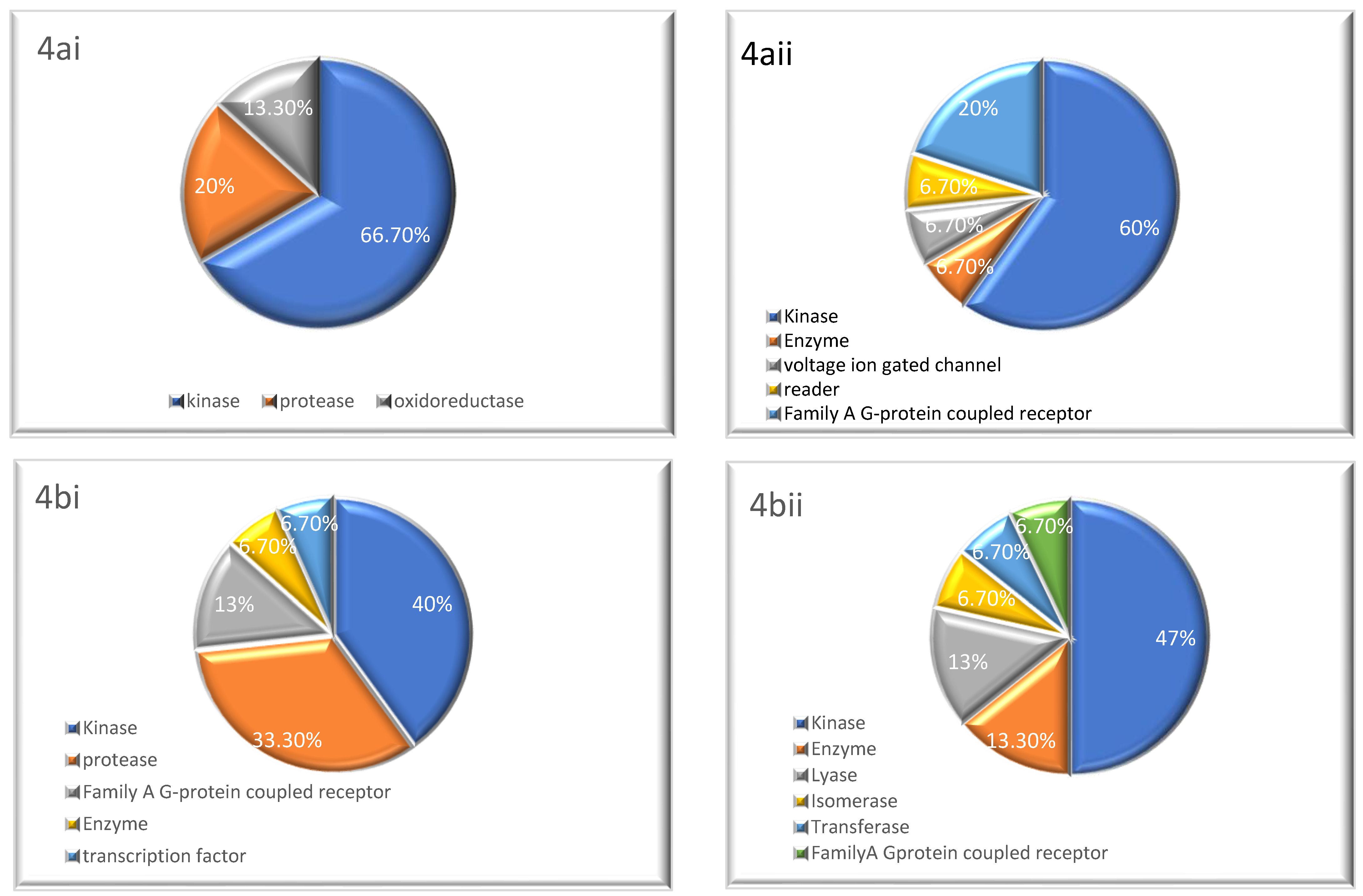
2.3. Target Prediction
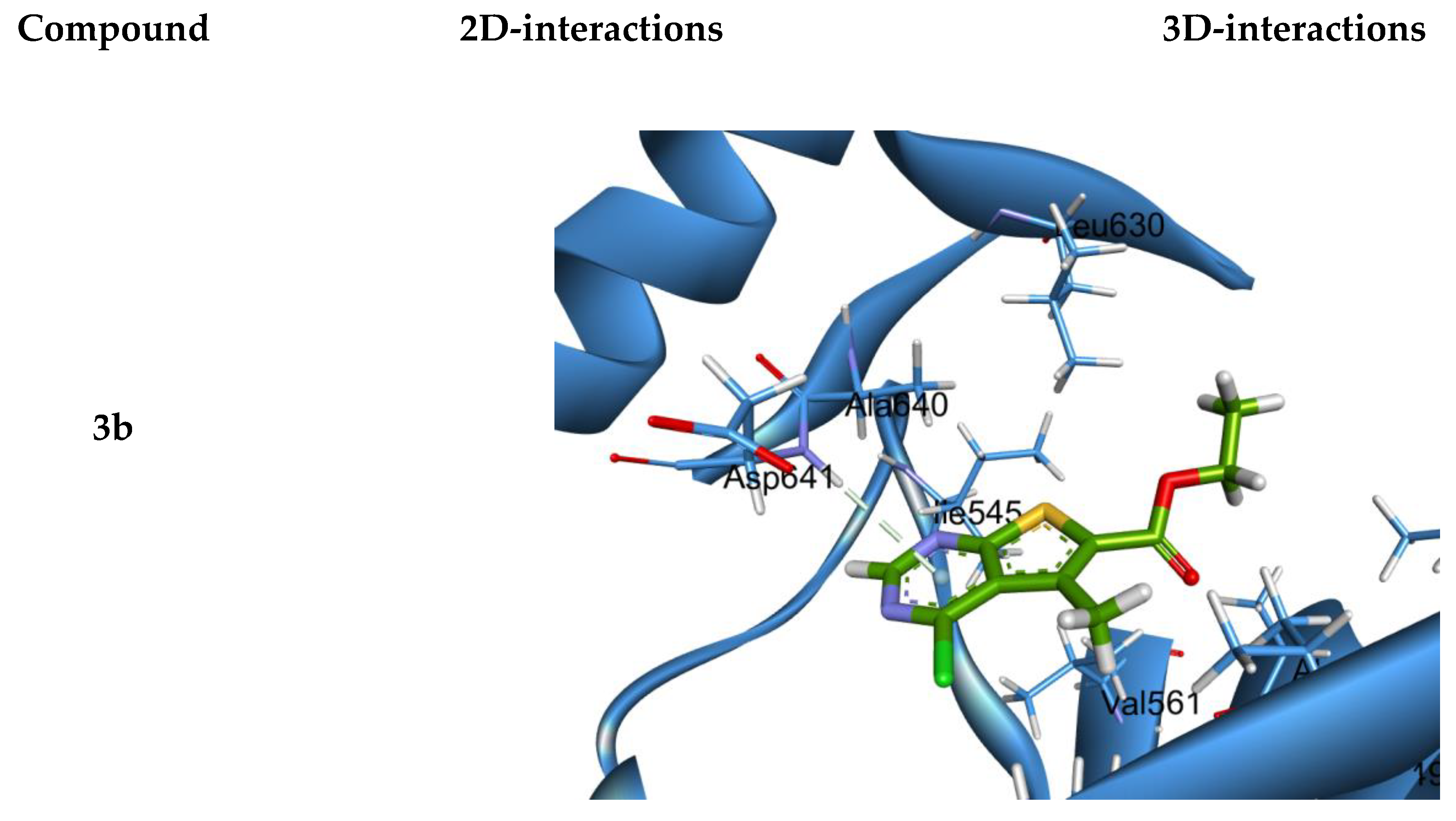
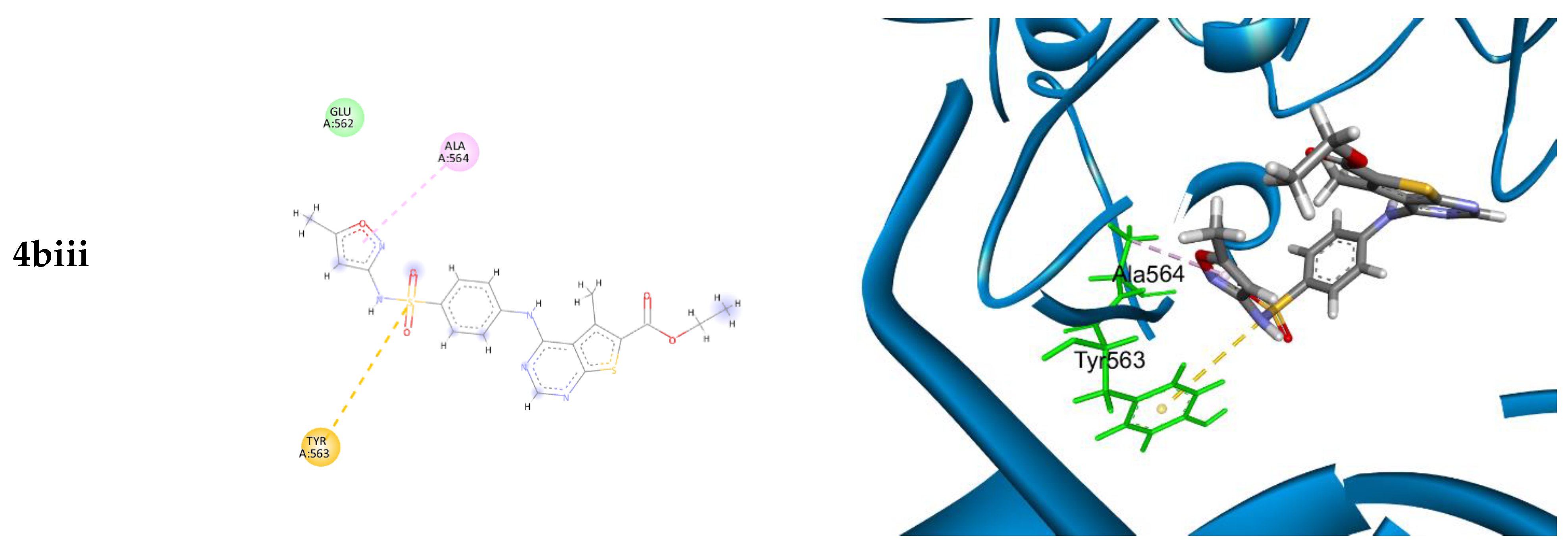
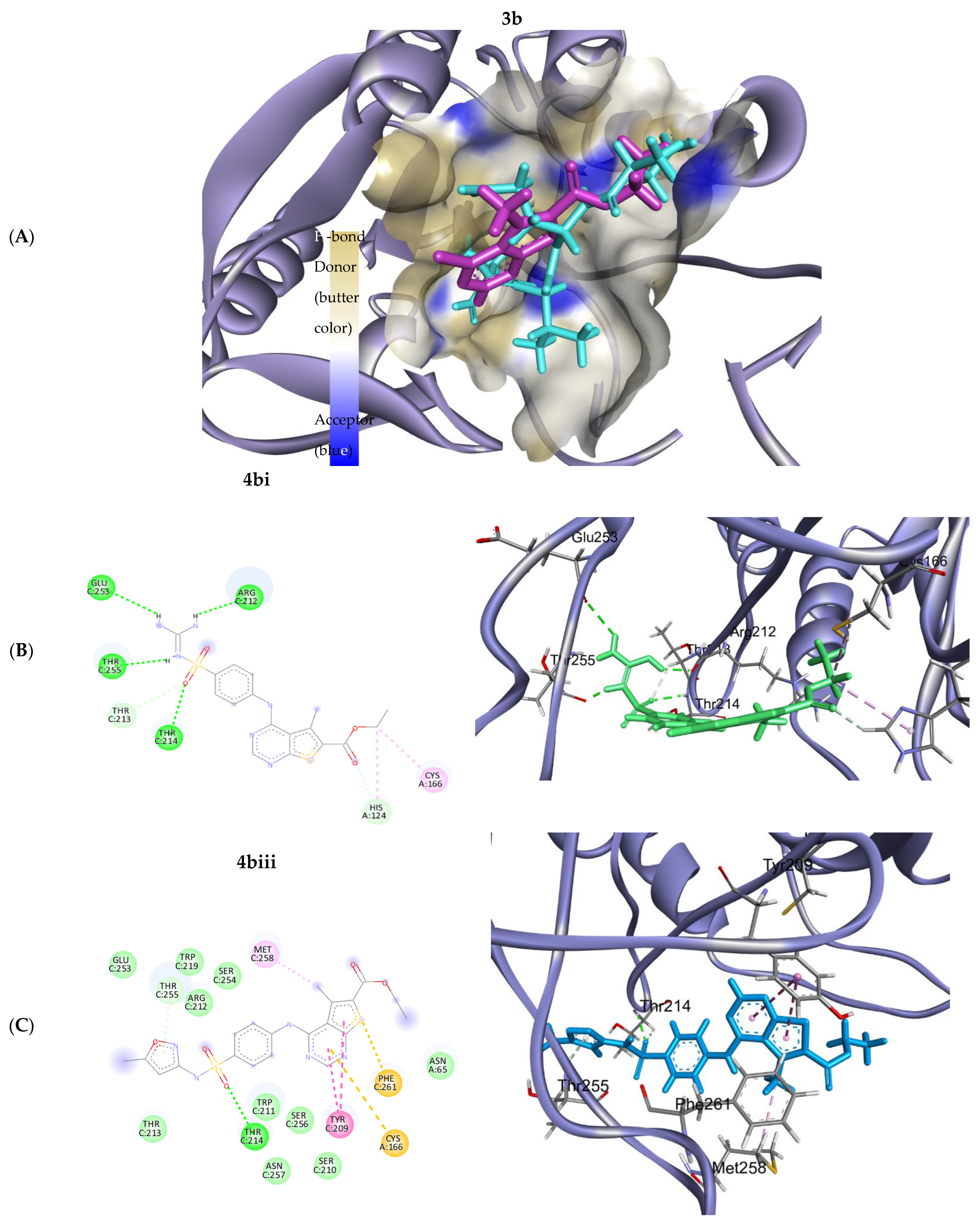
2.4. Modeling
2.5. Enzyme Specific Assay
2.5.1. FGFR-1 Inhibitory Activity
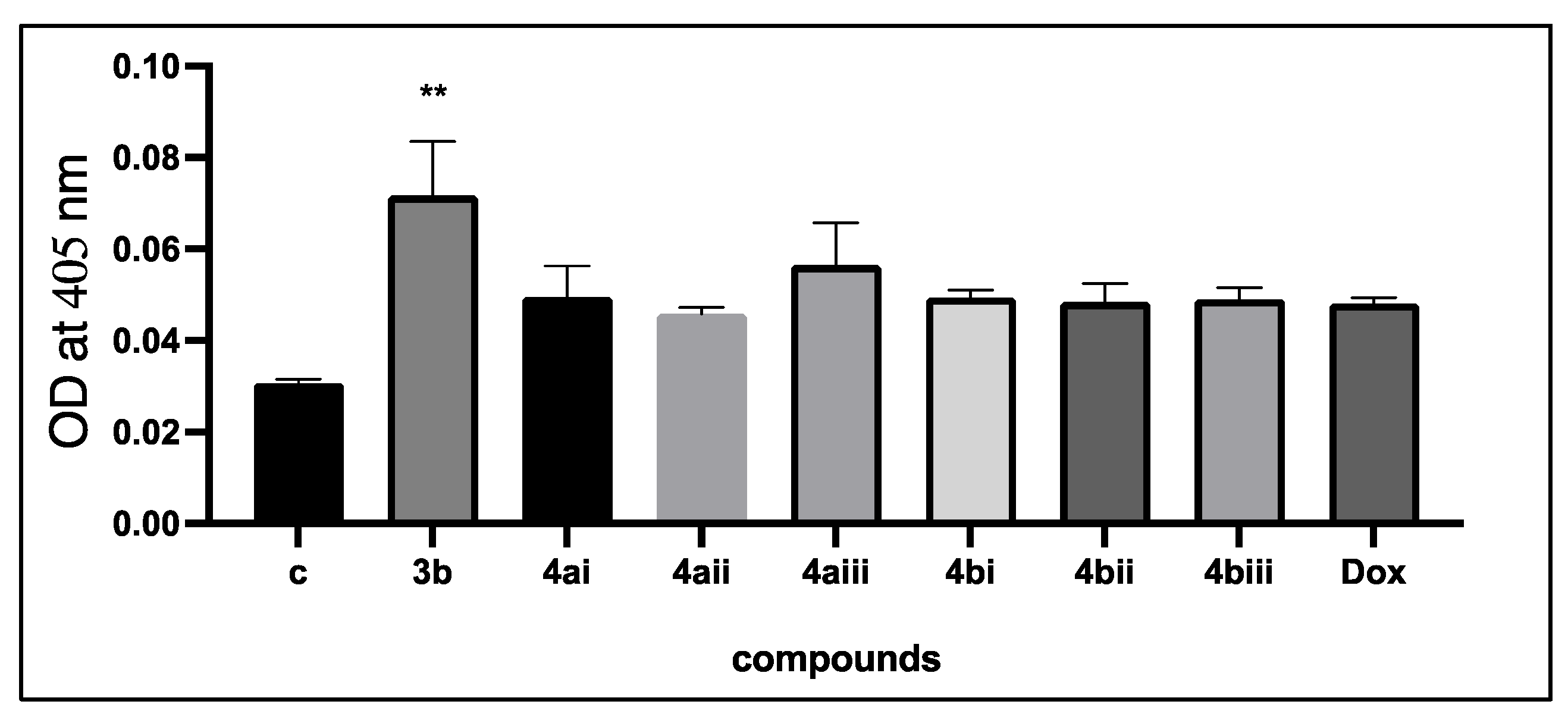
2.5.2. Caspase-3 Activity
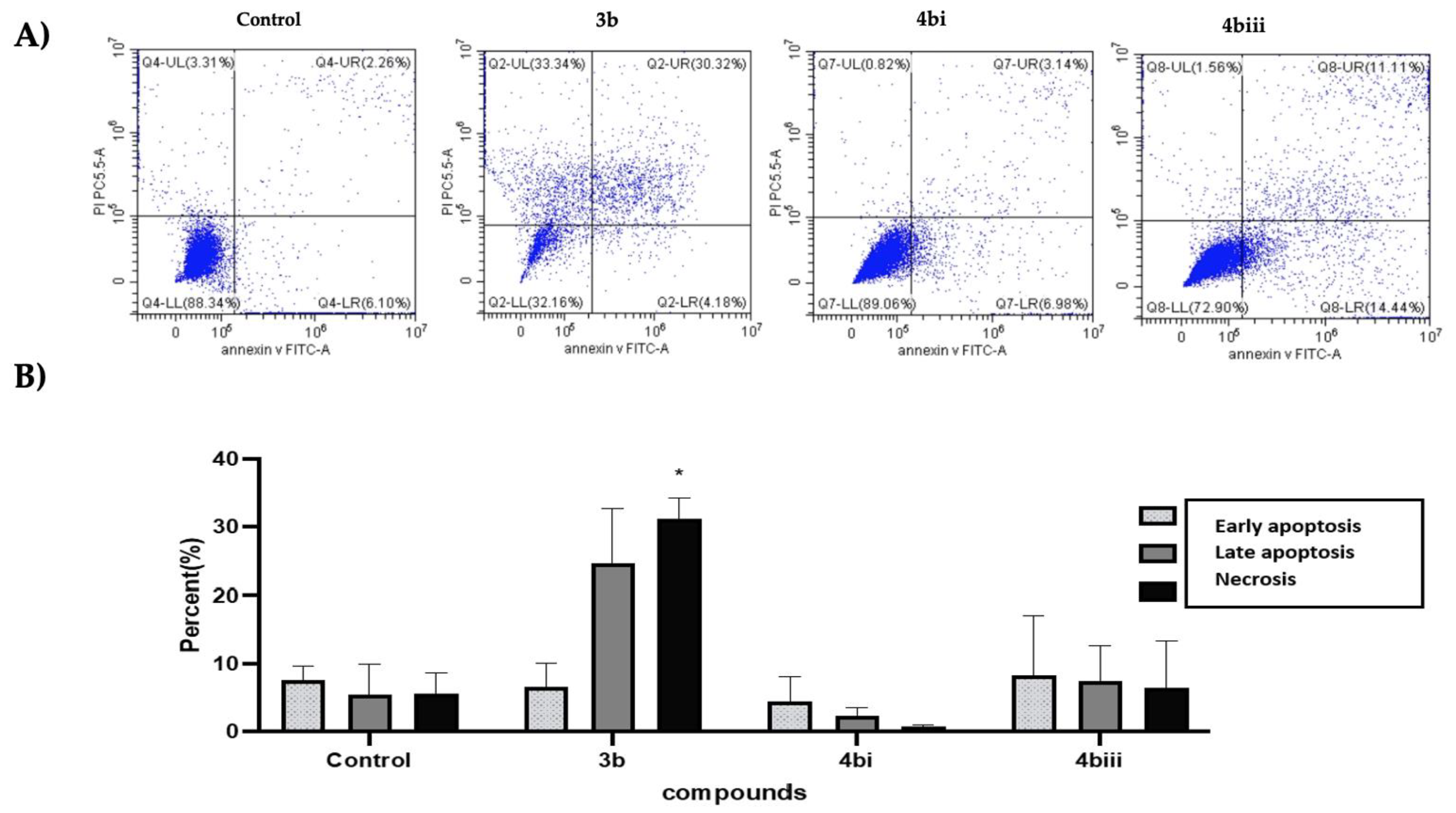
2.6. Annexin V/Propidium Iodide (PI) Flow Cytometric Analysis
2.7. Insilico Investigation
2.7.1. Investigation of Drug Likeness and Physicochemical Properties
2.7.2. Pharmacokinetics In Silico Assessment for the Prepared Hybrids
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. Anti-Proliferative Activities towards Breast Cancer Cells
3.2.2. Selectivity Index (SI)
3.2.3. FGFR-1 Inhibitory Activity
3.2.4. Caspase-3 Activity Assay
3.2.5. Evaluation of Apoptosis by Annexine V
3.3. Target Prediction
3.4. Molecular Modeling
3.5. Statistical Analysis
3.6. In Silico Drug Likeness and Pharmacokinetics Investigation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, F.; Wang, Y.; Nian, S.; Wang, Y.; Chen, D.; Yu, S.; Wang, S. Synthesis and evaluation of biological and antitumor activities of 5,7-dimethyl-oxazolo[5,4-d]pyrimidine-4,6(5H,7H)-dione derivatives as novel inhibitors of FGFR1. J. Enzym. Inhib. Med. Chem. 2015, 30, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wei, T.; Tang, Q.; Weng, B.; Li, W.; Jiang, X.; Ding, T.; Li, X.; Liang, G.; Cai, Y.; et al. Discovery and anticancer evaluation of two novel non-ATP-competitive FGFR1 inhibitors in non-small-cell lung cancer. BMC Cancer 2015, 15, 276. [Google Scholar] [CrossRef]
- Suneel Kumar, B.V.S.; Lakshmi, N.; Rambabu, G.; Raveendra, D.; Sarma, J.A.R.P. Fibroblast Growth Factor Receptor Inhibitors. Curr. Pharm. Des. 2012, 19, 687–701. [Google Scholar] [CrossRef]
- Bdzhola, V.G.; Yarmoluk, S.M.; Matyushok, V.I.; Balanda, A.O.; Yang, F.; Zhang, Y.; Ressler, S.J.; Ittmann, M.M.; Ayala, G.E.; Dang, T.D.; et al. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013, 73, 3716–3724. [Google Scholar]
- Gryshchenko, A.A.; Bdzhola, V.G.; Balanda, A.O.; Briukhovetska, N.V.; Kotey, I.M.; Golub, A.G.; Ruban, T.P.; Lukash, L.L.; Yarmoluk, S.M. Design, synthesis and biological evaluation of N-phenylthieno[2,3-d]pyrimidin-4-amines as inhibitors of FGFR1. Bioorg. Med. Chem. 2015, 23, 2287–2293. [Google Scholar] [CrossRef]
- Gryshchenko, A.A.; Bdzhola, V.G.; Pletnyova, L.V.; Chepurna, R.V.; Zhitnetsky, I.V.; Yarmoluk, S.M. Quinazolone inhibitors of protein kinase FGFR1. Ukr. Bioorg. Acta 2010, 8, 63–68. [Google Scholar]
- Gryshchenko, A.A.; Bdzhola, V.G.; Borovikov, O.V.; Kukharenko, O.P.; Pletnyova, L.V.; Yarmoluk, S.M. Search for FGFR1 inhibitors among oxindole derivatives. Ukr. Bioorg. Acta 2009, 7, 64–68. [Google Scholar]
- Elmongy, E.I. Thieno[2,3-d]pyrimidine derivatives: Synthetic approaches and their FLT3 kinase inhibition. J. Heterocycl. Chem. 2020, 57, 2067–2078. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Khedr, M.A.; Taleb, N.A.; Awad, H.M.; Abbas, S.E. Design, synthesis, and biological evaluation of some cyclohepta[b]thiophene and substituted pentahydrocycloheptathieno[2,3-d]pyrimidine derivatives. J. Heterocycl. Chem. 2017, 54, 1084–1093. [Google Scholar] [CrossRef]
- Sharaky, M.; Kamel, M.; Aziz, M.A.; Omran, M.; Rageh, M.M.; Abouzid, K.A.; Shouman, S.A. Design, synthesis and biological evaluation of a new thieno[2,3-d]pyrimidine-based urea derivative with potential antitumor activity against tamoxifen sensitive and resistant breast cancer cell lines. J. Enzym. Inhib. Med. Chem. 2020, 35, 1641–1656. [Google Scholar] [CrossRef]
- Abuelhassan, S.; Bakhite, E.A.G.; Abdel-Rahman, A.E.; El-Mahdy, A.F.M. Synthesis, characterization, and biological activities of some novel thienylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines and related heterocycles. J. Heterocycl. Chem. 2021, 58, 1784–1801. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Altwaijry, N.; Attallah, N.G.M.; AlKahtani, M.M.; Henidi, H.A. In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation. Pharmaceuticals 2022, 15, 170. [Google Scholar] [CrossRef]
- El-Mekabaty, A.; Fouda, A.E.; Shaaban, I. Convenient synthesis of functionalized thienopyrimidine-4-ones and thienopyridine-4-ones bearing a pyridine moiety with anticipated antioxidant activity. J. Heterocycl. Chem. 2020, 57, 2928–2935. [Google Scholar] [CrossRef]
- Nagaraju, K.; Bhaskaruni, V.H.S.S.; Kishore, R.; Maddila, S.; Singh, P.; Jonnalagadda, S.B. Synthesis and Antioxidant Evaluation of a New Class of Thienopyrimidine-rhodanine Hybrids. Lett. Drug Des. Discov. 2018, 15, 118126. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Khedr, M.A.; Taleb, N.A.; Abbas, S.E. Design and synthesis of new thienopyrimidine derivatives along with their antioxidant activity. Egypt. J. Chem. 2021, 64, 6857–6867. [Google Scholar] [CrossRef]
- Pingaew, R.; Prachayasittikul, V.; Worachartcheewan, A.; Thongnum, A.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Anticancer activity and QSAR study of sulfur-containing thiourea and sulfonamide derivatives. Heliyon 2022, 8, e10067. [Google Scholar] [CrossRef]
- Ali, E.M.H.; Abdel-Maksoud, M.S.; Oh, C. Thieno[2,3-d]pyrimidine as a promising scaffold in medicinal chemistry: Recent advances. Bioorg. Med. Chem 2019, 27, 1159–1194. [Google Scholar] [CrossRef]
- Wang, X.; Chen, D.; Yu, S.; Zhang, Z.; Wang, Y.; Qi, X.; Fu, W.; Xie, Z.; Ye, F. Synthesis and evaluation of biological and antitumor activities of tetrahydrobenzothieno[2,3-d]pyrimidine derivatives as novel inhibitors of FGFR-1. Chem. Biol. Drug Des. 2016, 87, 499–507. [Google Scholar] [CrossRef]
- Gryshchenko, A.A.; Levchenko, K.V.; Bdzhola, V.G.; Ruban, T.P.; Lukash, L.L.; Yarmoluk, S.M. Design, synthesis and biological evaluation of naphthostyril derivatives as novel protein kinase FGFR1 inhibitors. J. Enzym. Inhib. Med. Chem. 2015, 30, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Tarnavskiy, S.S.; Protopopov, M.V.; Borovykov, O.V.; Pryhodko, A.O.; Bdzhola, V.G.; Yarmoluk, S.M. Hit identification of FGFR-1 inhibitors using receptor-based virtual screening. Biopolym. Cell 2019, 35, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Elmongy, E.I.; Attallah, N.G.M.; Altwaijry, N.; AlKahtani, M.M.; Henidi, H.A. Design and synthesis of new thiophene/thieno[2,3-d]pyrimidines along with their cytotoxic biological evaluation as tyrosine kinase inhibitors in addition to their apoptotic and autophagic induction. Molecules 2021, 27, 123. [Google Scholar] [CrossRef]
- Hafez, H.N.; El-Gazzar, A.B.A. Design and synthesis of 3-pyrazolyl-thiophene, thieno[2,3-d]pyrimidines as new bioactive and pharmacological activities. Bioorg. Med. Chem. Lett. 2008, 18, 5222–5227. [Google Scholar] [CrossRef]
- Abdel-Atty, M.M.; Farag, N.A.; Serya, R.A.T.; Abouzid, K.A.M.; Mowafy, S. Molecular design, synthesis and in vitro biological evaluation of thienopyrimidine-hydroxamic acids as chimeric kinase HDAC inhibitors: A challenging approach to combat cancer. J. Enzym. Inhib. Med. Chem. 2021, 36, 1290–1311. [Google Scholar] [CrossRef]
- Wrobleski, S.T.; Wu, H.; Leftheris, K.; Das, J.; Hynes, J.; Lin, S. Inventorsphenyl-aniline substituted bicyclic compounds useful as kinase. inhibitors. Patent WO2005042537A1, 12 May 2005. [Google Scholar]
- Adel, M.; Serya, R.A.T.; Lasheen, D.S.; Abouzid, K.A.M. Identification of new pyrrolo[2,3-d]pyrimidines as potent VEGFR-2 tyrosine kinase inhibitors: Design, synthesis, biological evaluation and molecular modeling. Bioorg. Chem. 2018, 81, 612–629. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of in-vitro bioassay methods: Application in herbal drug research. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar]
- Mahavorasirikul, W.; Viyanant, V.; Chaijaroenkul, W.; Itharat, A.; Na-Bangchang, K. Cytotoxic activity of thai medicinal plants against human cholangiocarcinoma, laryngeal and hepatocarcinoma cells in vitro. BMC Complement. Altern. Med. 2010, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood–Brain barrier (BBB) score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Elmongy, E.; Henidi, H. In silico evaluation of a promising key intermediate thieno [2,3-d] pyrimidine derivative with expected JAK2 kinase inhibitory activity. MolBank 2022, 2022, M1352. [Google Scholar] [CrossRef]
- Wu, C.; Coumar, M.S.; Chu, C.; Lin, W.-H.; Chen, Y.-R.; Chen, C.-T.; Shiao, H.-Y.; Rafi, S.; Wang, S.-Y.; Hsu, H.; et al. Design and synthesis of tetrahydropyridothieno[2,3-d]pyrimidine scaffold based epidermal growth factor receptor (EGFR) kinase inhibitors: The role of side chain chirality and michael acceptor group for maximal potency. J. Med. Chem. 2010, 53, 7316–7326. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Gewald, K.; Schinke, E.; Böttcher, H. Heterocyclen aus CH-aciden nitrilen, VIII. 2-amino-thiophene aus methylenaktiven nitrilen, carbonylverbindungen und schwefel. Chem. Ber. 1966, 99, 94–100. [Google Scholar] [CrossRef]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General cytotoxicity assessment by means of the MTT assay. In Methods in Molecular Biology (Clifton, N.J.); Springer: New York, NY, USA, 2015; Volume 1250, pp. 333–348. [Google Scholar]
- Pagano, K.; Torella, R.; Foglieni, C.; Bugatti, A.; Tomaselli, S.; Zetta, L.; Presta, M.; Rusnati, M.; Taraboletti, G.; Colombo, G.; et al. Direct and allosteric inhibition of the FGF2/HSPGs/FGFR-1 ternary complex formation by an antiangiogenic, thrombospondin-1-mimic small molecule. PLoS ONE. 2012, 7, e36990. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2021. [Google Scholar]
- Protein Data Bank. Available online: https://www.rcsb.org/structure/5o49 (accessed on 10 January 2022).
- Protein Data Bank. Available online: https://www.rcsb.org/structure/7JL7 (accessed on 18 January 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 * (µM) | ||
|---|---|---|---|
| MCF-7 | MDA-MB-231 | MCF-10A (Non-Tumorigenic Cells) | |
| 3b | 9.74 ± 0.13 | 4.45 ± 0.31 | 264.9 ± 4.2 |
| 4ai | 35.79 ± 1.2 | 40.17 ± 2.7 | 457.9 ± 12.3 |
| 4aii | 22.29 ± 3.1 | 20.19 ± 1.5 | >1000 |
| 4aiii | 67.07 ± 15.7 | 68.345 ± 4.6 | 88.45 ± 10.1 |
| 4bi | 6.17 ± 1.3 | 8.68 ± 4.3 | 277.05 ± 7.9 |
| 4bii | 13.97 ± 8.2 | 15.57 ± 0.1 | 229.9 ± 24.1 |
| 4biii | 9.54 ± 3.1 | 17.64 ± 2.3 | 244.5 ± 1.9 |
| Doxorubicin | 1.6 ± 2.2 | 2.2 ± 0.3 | 2.8 ± 1.2 |
| Compound No. | MCF-7 | MDA-MB231 |
|---|---|---|
| 3b | 27.20 | 59.45 |
| 4ai | 12.79 | 11.40 |
| 4aii | ND a | ND a |
| 4aiii | 1.32 | 1.29 |
| 4bi | 44.86 | 31.90 |
| 4bii | 16.45 | 14.77 |
| 4biii | 25.62 | 13.86 |
| Doxorubicin | 1.75 | 1.27 |
| Compound | Protein-Pdb Code | Binding Energy | RMSD | Amino Acids Involved in Interaction | Types of Interaction |
|---|---|---|---|---|---|
| 3b | 5O49 | −5.239 | 1.0171 | ASP 641 | Pi-H |
| 4ai | 5O49 | −6.866 | 1.873 | GLU 531 LEU 484 GLY 567 | H-donor Pi-H Pi-H |
| 4aii | 5O49 | −5.989 | 1.905 | ASP 641 LYS 514 GLY 567 | H-donor H-acceptor Pi-H |
| 4aiii | 5O49 | −7.091 | 2.191 | ALA 564 | H-acceptor |
| 4bi | 5O49 | −6.443 | 2.093 | ASP 641 TYR 563 LEU 484 | H-donor H-acceptor Pi-H |
| 4bii | 5O49 | −6.365 | 1.781 | ASP 641 TYR 563 LEU 484 LYS 514 | H-acceptor H-acceptor Pi-H Pi-cation |
| 4biii | 5O49 | −7.123 | 1.776 | ALA 564 TYR 563 | Pi-H Pi-sulfur |
| Compound | Protein Pdb Code | Binding Energy | RMSD | Amino Acids Involved in Interaction | Types of Interaction |
|---|---|---|---|---|---|
| 3b | 7JL7 | −4.059 | 1.7578 | ARG 212 | H-donor |
| 4ai | 7JL7 | −6.157 | 1.372 | SER 123 TYR 209 TYR 209 ARG 67 | H-donor H-pi H-pi Pi-cation |
| 4aii | 7JL7 | −6.245 | 2.455 | ARG212 | H-acceptor |
| 4aiii | 7JL7 | −5.803 | 2.128 | THR 255 | H-acceptor |
| 4bi | 7JL7 | −5.925 | 2.166 | THR 255 THR 214 ARG212 GLU 253 CYS 166 HIS 124 | H-acceptor H-donor H-bond H-bond H-bond Pi-H |
| 4bii | 7JL7 | −4.981 | 1.978 | PRO 66 ASN 257 ASN 65 | H-donor H-acceptor Pi-H |
| 4biii | 7JL7 | −5.939 | 1.801 | THR 214 TYR 209 MET 258 CYS 166 PHE 261 | H-donor π-π π-π π-sulfur π-sulfur |
| Compound No. | IC50 (μM) |
|---|---|
| 3b | 179.15 ± 5.6 |
| 4ai | 131.95 ± 4.3 |
| 4aii | 119.43 ± 5.6 |
| 4aiii | 159.2 ± 5.3 |
| 4bi | 325.65 ± 7.4 |
| 4bii | 289.166 ± 12.0 |
| 4biii | 168.05 ± 8.3 |
| Doxorubicin | 53.09 ± 3.2 |
| MW | HBA | HBD | MR | TPSA | iLog p | Lipinski Violations | Drug Likeness | |
|---|---|---|---|---|---|---|---|---|
| 3b | 256.71 | 4 | 0 | 63.48 | 80.32 | 2.6 | 0 | −0.34 |
| 4ai | 402.49 | 5 | 4 | 106.58 | 170.47 | 1.51 | 0 | 1.58 |
| 4aii | 438.53 | 6 | 2 | 117.09 | 146.38 | 2.66 | 0 | 1.03 |
| 4aiii | 441.53 | 6 | 2 | 116.53 | 146.63 | 2.59 | 0 | 1.08 |
| 4bi | 434.49 | 7 | 4 | 110.21 | 196.77 | 1.75 | 0 | 1.19 |
| 4bii | 470.52 | 8 | 2 | 120.71 | 172.68 | 2.78 | 0 | 0.88 |
| 4biii | 473.53 | 8 | 2 | 120.15 | 172.93 | 3.1 | 0 | 0.73 |
| Cpd. No. | ABSORPTION | ||||
| Water solubility (log mol/L) | Caco2 permeability * | Intestinal absorption (human)% | log Kp ** | ||
| 3b | −3.458 | 1.324 | 96.254 | −2.895 | |
| 4ai | −3.037 | 0.207 | 77.346 | −2.735 | |
| 4aii | −4.457 | 0.838 | 88.833 | −2.892 | |
| 4aiii | −4.743 | 0.72 | 95.347 | −2.901 | |
| 4bi | −3.004 | −0.261 | 64.447 | −2.735 | |
| 4bii | −4.108 | 0.329 | 75.934 | −2.85 | |
| 4biii | −4.387 | 0.211 | 82.448 | −2.871 | |
| Cpd. No. | DISTRIBUTION | EXCRETION | TOXICITY | ||
| VDss *** (human) | BBB **** permeability | CNS permeability | Total Clearance | Max. tolerated dose (human) | |
| 3b | −0.359 | 0.184 | −2.91 | 0.337 | 0.84 |
| 4ai | 0.397 | −0.876 | −2.655 | −0.07 | 0.829 |
| 4aii | 0.137 | −0.906 | −2.309 | −0.097 | −0.546 |
| 4aiii | 0.176 | −0.852 | −2.182 | −0.071 | −0.611 |
| 4bi | 0.372 | −1.323 | −3.195 | −0.107 | 0.602 |
| 4bii | −0.277 | −1.583 | −3.249 | 0.031 | −0.561 |
| 4biii | −0.27 | −1.529 | −2.722 | 0.057 | −0.612 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmongy, E.I.; Binjubair, F.A.; Alshehri, O.Y.; Baeshen, K.A.; Almukhalfi, Z.A.; Henidi, H.A. In Silico Screening and Anticancer-Apoptotic Evaluation of Newly Synthesized Thienopyrimidine/Sulfonamide Hybrids. Int. J. Mol. Sci. 2023, 24, 10827. https://doi.org/10.3390/ijms241310827
Elmongy EI, Binjubair FA, Alshehri OY, Baeshen KA, Almukhalfi ZA, Henidi HA. In Silico Screening and Anticancer-Apoptotic Evaluation of Newly Synthesized Thienopyrimidine/Sulfonamide Hybrids. International Journal of Molecular Sciences. 2023; 24(13):10827. https://doi.org/10.3390/ijms241310827
Chicago/Turabian StyleElmongy, Elshaymaa I., Faizah A. Binjubair, Ohoud Y. Alshehri, Kholoud A. Baeshen, Zaha A. Almukhalfi, and Hanan A. Henidi. 2023. "In Silico Screening and Anticancer-Apoptotic Evaluation of Newly Synthesized Thienopyrimidine/Sulfonamide Hybrids" International Journal of Molecular Sciences 24, no. 13: 10827. https://doi.org/10.3390/ijms241310827