
Exploring the Role of Enhancer-Mediated Transcriptional Regulation in Precision Biology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
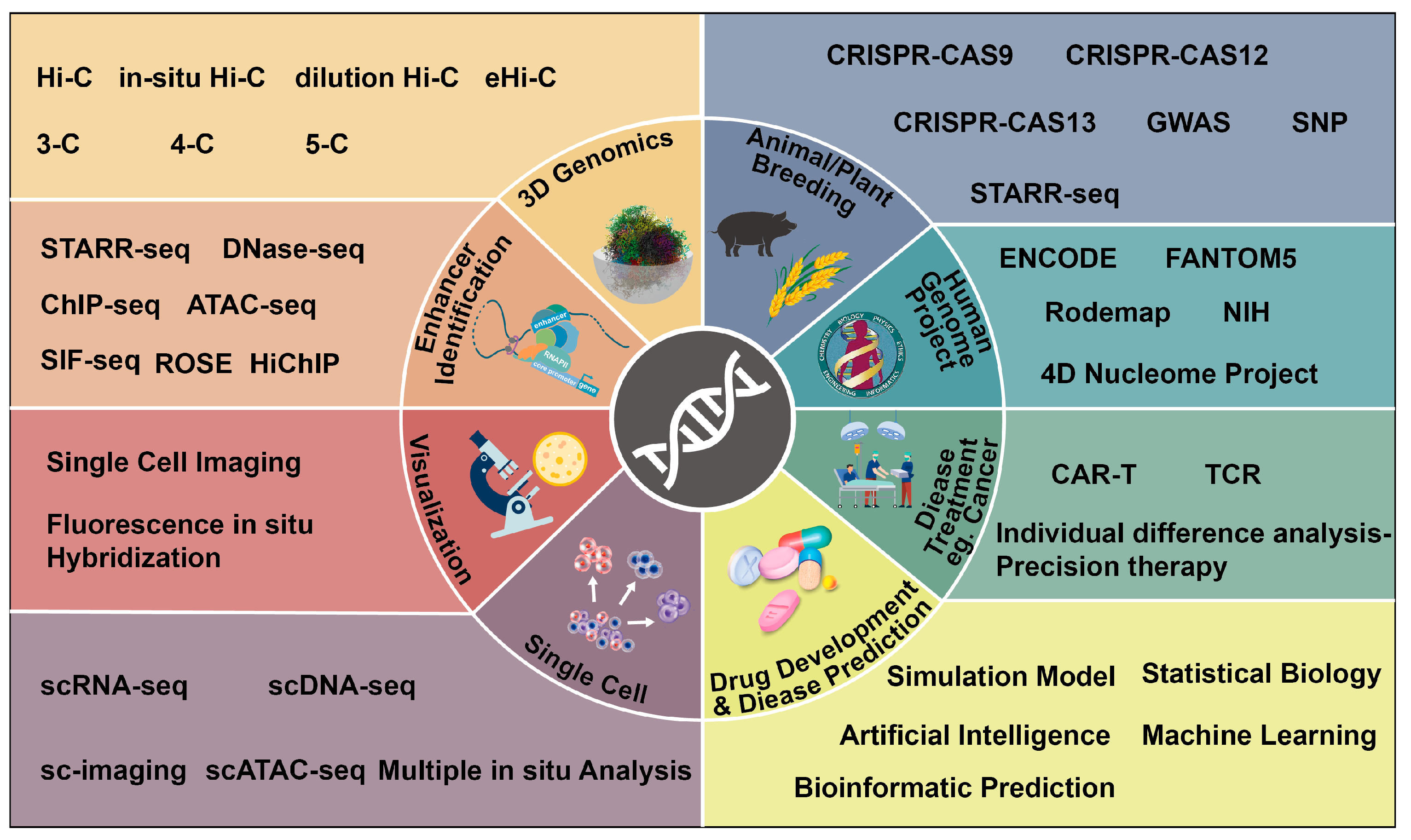
:1. Introduction
2. The Development of Precision Biology
3. Transcriptional Regulation and Precision Biology
3.1. Transcription Factors and Transcriptional Apparatus
3.2. Transcriptional Regulatory Elements
3.3. Regulation of Chromatin Dynamics in Transcription
4. The Role of Different Enhancers in Transcriptional Regulation
4.1. Typical Enhancers
4.2. Annotation of Typical Enhancers
4.3. Super Enhancer
4.3.1. Comparison of Typical Enhancers and Super Enhancers
4.3.2. Annotation of Super Enhancers
4.3.3. Regulatory Network of Super Enhancers
4.4. Multi-Enhancer Modulation
4.4.1. Multi-Enhancer Modulation Affects Biological Effects
4.4.2. Complex Molecular Mechanisms of Multi-Enhancer Regulation
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weil, A.R. Precision Medicine. Health Aff. 2018, 37, 687. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Precision medicine in nonalcoholic fatty liver disease: New therapeutic insights from genetics and systems biology. Clin. Mol. Hepatol. 2020, 26, 461–475. [Google Scholar] [CrossRef]
- Fountzilas, E.; Tsimberidou, A.M.; Vo, H.H.; Kurzrock, R. Clinical trial design in the era of precision medicine. Genome Med. 2022, 14, 101. [Google Scholar] [CrossRef]
- Alaimo, S.; Giugno, R.; Acunzo, M.; Veneziano, D.; Ferro, A.; Pulvirenti, A. Post-transcriptional knowledge in pathway analysis increases the accuracy of phenotypes classification. Oncotarget 2016, 7, 54572–54582. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Galis, Z.S. Editorial: Where Is Waldo: Contextualizing the Endothelial Cell in the Era of Precision Biology. Front. Cardiovasc. Med. 2020, 7, 127. [Google Scholar] [CrossRef]
- Stadhouders, R.; Filion, G.J.; Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 2019, 569, 345–354. [Google Scholar] [CrossRef]
- Bentovim, L.; Harden, T.; DePace, A.H. Transcriptional precision and accuracy in development: From measurements to models and mechanisms. Development 2017, 144, 3855–3866. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wong, C.-H.; Birnbaum, R.Y.; Li, G.; Favaro, R.; Ngan, C.Y.; Lim, J.; Tai, E.; Poh, H.M.; Wong, E.; et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 2013, 504, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Nagai, S.; Davis, R.E.; Mattei, P.J.; Eagen, K.P.; Kornberg, R.D. Chromatin potentiates transcription. Proc. Natl. Acad. Sci. USA 2017, 114, 1536–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvon, E.Z.; Kazmar, T.; Stampfel, G.; Yáñez-Cuna, J.O.; Pagani, M.; Schernhuber, K.; Dickson, B.J.; Stark, A. Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 2014, 512, 91–95. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Laiker, I.; Frankel, N. Pleiotropic Enhancers are Ubiquitous Regulatory Elements in the Human Genome. Genome Biol. Evol. 2022, 14, evac071. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-T.; Shah, R.H.; Tegay, D.; Onel, K. Precision oncology: Lessons learned and challenges for the future. Cancer Manag. Res. 2019, 11, 7525–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Schuijers, J.; Lin, C.Y. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liang, H. A High-Resolution Map of Human Enhancer RNA Loci Characterizes Super-enhancer Activities in Cancer. Cancer Cell 2020, 38, 701–715. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, X.; Li, Q.; Kong, D.; Huang, Q.; Luo, J.; Kong, S.; Peng, Y.; Zhang, Y. Distinct enhancer-promoter modes determine Sox 2 regulation in mouse pluripotent cells. Genes Dis. 2022. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Auffray, C.; Griffin, J.L.; Khoury, M.J.; Lupski, J.R.; Schwab, M. Ten years of Genome Medicine. Genome Med. 2019, 11, 7. [Google Scholar] [CrossRef]
- Goetz, L.H.; Schork, N.J. Personalized medicine: Motivation, challenges, and progress. Fertil. Steril. 2018, 109, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Elemento, O. The future of precision medicine: Towards a more predictive personalized medicine. Emerg. Top. Life Sci. 2020, 4, 175–177. [Google Scholar] [CrossRef]
- Ozdemir, E.S.; Ranganathan, S.V.; Nussinov, R. How has artificial intelligence impacted COVID-19 drug repurposing and what lessons have we learned? Expert Opin. Drug Discov. 2022, 17, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Filipp, F.V. Opportunities for Artificial Intelligence in Advancing Precision Medicine. Curr. Genet. Med. Rep. 2019, 7, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Lee, J.; Zhang, S.; Benyshek, C.; Dokmeci, M.R.; Khademhosseini, A. Engineering Precision Medicine. Adv. Sci. 2019, 6, 1801039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Splinter, E.; de Laat, W. The complex transcription regulatory landscape of our genome: Control in three dimensions. EMBO J. 2011, 30, 4345–4355. [Google Scholar] [CrossRef] [Green Version]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.; Li, Q.; Zhang, G.; Li, Q.; Huang, Q.; Huang, L.; Zhang, H.; Huang, Y.; Peng, Y.; Qin, B.; et al. Exonuclease combinations reduce noises in 3D genomics technologies. Nucleic Acids Res. 2020, 48, e44. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Fullwood, M.J.; Xu, H.; Mulawadi, F.H.; Velkov, S.; Vega, V.; Ariyaratne, P.N.; Bin Mohamed, Y.; Ooi, H.-S.; Tennakoon, C.; et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010, 11, R22. [Google Scholar] [CrossRef] [Green Version]
- Solomon, M.J.; Larsen, P.L.; Varshavsky, A. Mapping protein-DNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell 1988, 53, 937–947. [Google Scholar] [CrossRef]
- Mundade, R.; Ozer, H.G.; Wei, H.; Prabhu, L.; Lu, T. Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond. Cell Cycle 2014, 13, 2847–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Li, C.; Chen, Z.; Zhang, Y.; Tian, Q.; Sun, M.; Zhang, S.; Yu, M.; Wang, G. An intellectual disability-related MED23 mutation dysregulates gene expression by altering chromatin conformation and enhancer activities. Nucleic Acids Res. 2023, 51, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Xu, W.; Hong, P.; Wu, C.; Zhang, Z.; Zhang, S.; Xing, L.; Yang, B.; Zhou, W.; Xiao, Q.; et al. Decoding the spatial chromatin organization and dynamic epigenetic landscapes of macrophage cells during differentiation and immune activation. Nat. Commun. 2022, 13, 5857. [Google Scholar] [CrossRef] [PubMed]
- Dickel, D.E.; Zhu, Y.; Nord, A.S.; Wylie, J.N.; Akiyama, J.A.; Afzal, V.; Plajzer-Frick, I.; Kirkpatrick, A.; Göttgens, B.; Bruneau, B.G.; et al. Function-based identification of mammalian enhancers using site-specific integration. Nat. Methods 2014, 11, 566–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, Y.; Zhang, Q.; Wu, J.; Liang, J.; Yu, S.; Wei, G.-H.; White, K.P.; Wang, X. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017, 18, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit, M. Shooting for the STARRs: A Modified STARR-seq Assay for Rapid Identification and Evaluation of Plant Regulatory Sequences in Tobacco Leaves. Plant Cell 2020, 32, 2057–2058. [Google Scholar] [CrossRef] [PubMed]
- Sbalzarini, I.F.; Koumoutsakos, P. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 2005, 151, 182–195. [Google Scholar] [CrossRef]
- Lee, L.P. Satellite nanoscope and cellular BioASICs for quantitative biomedicine. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2009, 2009, 4582–4585. [Google Scholar] [CrossRef]
- Dasgupta, A.; Deschamps, J.; Matti, U.; Hübner, U.; Becker, J.; Strauss, S.; Jungmann, R.; Heintzmann, R.; Ries, J. Direct supercritical angle localization microscopy for nanometer 3D superresolution. Nat. Commun. 2021, 12, 1180. [Google Scholar] [CrossRef]
- Tanay, A.; Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 2017, 541, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chessel, A.; Carazo Salas, R.E. From observing to predicting single-cell structure and function with high-throughput/high-content microscopy. Essays Biochem. 2019, 63, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Ang, C.E.; Zhuang, X. Spatially resolved epigenomic profiling of single cells in complex tissues. Cell 2022, 185, 4448–4464. [Google Scholar] [CrossRef]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Shema, E.; Bernstein, B.E.; Buenrostro, J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019, 51, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chin, C.R.; Ying, H.Y.; Meydan, C.; Teater, M.R.; Xia, M.; Farinha, P.; Takata, K.; Chu, C.S.; Rivas, M.A.; et al. Cooperative super-enhancer inactivation caused by heterozygous loss of CREBBP and KMT2D skews B cell fate decisions and yields T cell-depleted lymphomas. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Caliskan, M.; Billstrand, C.; Michelini, K.; Chavarria, C.; De Leon, S.; Mitrano, A.; Lewellyn, N.; Elias, J.A.; Chupp, G.L.; et al. Integrated analyses of gene expression and genetic association studies in a founder population. Hum. Mol. Genet. 2016, 25, 2104–2112. [Google Scholar] [CrossRef] [Green Version]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef]
- Zhou, X.; Lu, Y.; Zhao, F.; Dong, J.; Ma, W.; Zhong, S.; Wang, M.; Wang, B.; Zhao, Y.; Shi, Y.; et al. Deciphering the spatial-temporal transcriptional landscape of human hypothalamus development. Cell Stem Cell 2022, 29, 328–343. [Google Scholar] [CrossRef]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, G.; Zhou, J.; Xu, R.; Lu, Q.; Wang, H. Cross-Cell-Type Prediction of TF-Binding Site by Integrating Convolutional Neural Network and Adversarial Network. Int. J. Mol. Sci. 2019, 20, 3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnung, L.; Vos, S.M. Assembly of RNA polymerase II transcription initiation complexes. Curr. Opin. Struct. Biol. 2022, 73, 102335. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Biddick, R.; Young, E.T. Yeast Mediator and its role in transcriptional regulation. Comptes Rendus Biol. 2005, 328, 773–782. [Google Scholar] [CrossRef]
- Kelleher, R.J., 3rd; Flanagan, P.M.; Kornberg, R.D. A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 1990, 61, 1209–1215. [Google Scholar] [CrossRef]
- Flanagan, P.M.; Kelleher, R.J., 3rd; Sayre, M.H.; Tschochner, H.; Kornberg, R.D. A mediator required for activation of RNA polymerase II transcription in vitro. Nature 1991, 350, 436–438. [Google Scholar] [CrossRef]
- Gabriele, M.; Brandão, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.-H.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef]
- Abdella, R.; Talyzina, A.; Chen, S.; Inouye, C.J.; Tjian, R.; He, Y. Structure of the human Mediator-bound transcription preinitiation complex. Science 2021, 372, 52–56. [Google Scholar] [CrossRef]
- Rengachari, S.; Schilbach, S.; Aibara, S.; Dienemann, C.; Cramer, P. Structure of the human Mediator-RNA polymerase II pre-initiation complex. Nature 2021, 594, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qi, Y.; Wu, Z.; Wang, X.; Li, J.; Zhao, D.; Hou, H.; Li, Y.; Yu, Z.; Liu, W.; et al. Structural insights into preinitiation complex assembly on core promoters. Science 2021, 372, eaba8490. [Google Scholar] [CrossRef]
- Xu, M.; Gonzalez-Hurtado, E.; Martinez, E. Core promoter-specific gene regulation: TATA box selectivity and Initiator-dependent bi-directionality of serum response factor-activated transcription. Biochim. Biophys. Acta 2016, 1859, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Chen, K.; Zhang, W.; Chen, Z. Structure of human chromatin-remodelling PBAF complex bound to a nucleosome. Nature 2022, 605, 166–171. [Google Scholar] [CrossRef]
- Kyrchanova, O.; Georgiev, P. Mechanisms of Enhancer-Promoter Interactions in Higher Eukaryotes. Int. J. Mol. Sci. 2021, 22, 671. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, T.L.; Hossain, A.; Salis, H.M. Automated model-predictive design of synthetic promoters to control transcriptional profiles in bacteria. Nat. Commun. 2022, 13, 5159. [Google Scholar] [CrossRef] [PubMed]
- Melancon, P.; Burgess, R.R.; Record, M.T., Jr. Nitrocellulose filter binding studies of the interactions of Escherichia coli RNA polymerase holoenzyme with deoxyribonucleic acid restriction fragments: Evidence for multiple classes of nonpromoter interactions, some of which display promoter-like properties. Biochemistry 1982, 21, 4318–4331. [Google Scholar] [CrossRef]
- Melançon, P.; Burgess, R.R.; Record, M.T., Jr. Direct evidence for the preferential binding of Escherichia coli RNA polymerase holoenzyme to the ends of deoxyribonucleic acid restriction fragments. Biochemistry 1983, 22, 5169–5176. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Reiter, F.; Wienerroither, S.; Stark, A. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017, 43, 73–81. [Google Scholar] [CrossRef]
- Rickels, R.; Shilatifard, A. Enhancer Logic and Mechanics in Development and Disease. Trends Cell Biol. 2018, 28, 608–630. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Das, N.D.; Umehara, T.; Koseki, H. Factors and Mechanisms That Influence Chromatin-Mediated Enhancer-Promoter Interactions and Transcriptional Regulation. Cancers 2022, 14, 5404. [Google Scholar] [CrossRef] [PubMed]
- Sungalee, S.; Liu, Y.; Lambuta, R.A.; Katanayeva, N.; Collier, M.D.; Tavernari, D.; Roulland, S.; Ciriello, G.; Oricchio, E. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat. Genet. 2021, 53, 650–662. [Google Scholar] [CrossRef]
- Ramirez, R.N.; Chowdhary, K.; Leon, J.; Mathis, D.; Benoist, C. FoxP3 associates with enhancer-promoter loops to regulate T(reg)-specific gene expression. Sci. Immunol. 2022, 7, eabj9836. [Google Scholar] [CrossRef] [PubMed]
- Crump, N.T.; Ballabio, E.; Godfrey, L.; Thorne, R.; Repapi, E.; Kerry, J.; Tapia, M.; Hua, P.; Lagerholm, C.; Filippakopoulos, P.; et al. BET inhibition disrupts transcription but retains enhancer-promoter contact. Nat. Commun. 2021, 12, 223. [Google Scholar] [CrossRef] [PubMed]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef] [Green Version]
- De Laat, W.; Duboule, D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 2013, 502, 499–506. [Google Scholar] [CrossRef]
- Gregor, T.; Garcia, H.G.; Little, S.C. The embryo as a laboratory: Quantifying transcription in Drosophila. Trends Genet. 2014, 30, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Liang, P.; Long, C.; Li, H.; Li, H.; Liang, Y.; He, X.; Xi, Q.; Xing, Y.; Zuo, Y. EmAtlas: A comprehensive atlas for exploring spatiotemporal activation in mammalian embryogenesis. Nucleic Acids Res. 2023, 51, 924–932. [Google Scholar] [CrossRef]
- Adhya, S. Multipartite genetic control elements: Communication by DNA loop. Annu. Rev. Genet. 1989, 23, 227–250. [Google Scholar] [CrossRef]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Banerji, J.; Rusconi, S.; Schaffner, W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 1981, 27, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Hen, R.; Wasylyk, B.; Everett, R.; Gaub, M.; Chambon, P. The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res. 1981, 9, 6047–6068. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef]
- Rivera, C.M.; Ren, B. Mapping human epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef] [Green Version]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Kenneth Baillie, J.; de Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.M.; de la Torre, V.; Richardson, D.; Royo, R.; Puiggròs, M.; Moncunill, V.; Fragkogianni, S.; Clarke, L.; Flicek, P.; Rico, D.; et al. The BLUEPRINT Data Analysis Portal. Cell Syst. 2016, 3, 491–495.e495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser–A database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; He, B.; Liu, S.; Zhu, H.; Tan, K.; Qian, J. EnhancerAtlas: A resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics 2016, 32, 3543–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, Q.; Zhang, W.; Lin, J.-R.; Cai, Y.; Mitra, J.; Zhang, Z.D. HEDD: Human Enhancer Disease Database. Nucleic Acids Res. 2018, 46, D113–D120. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Bonn, S.; Zinzen, R.; Girardot, C.; Gustafson, E.H.; Gonzalez, A.P.; Delhomme, N.; Ghavi-Helm, Y.; Wilczynski, B.; Riddell, A.; Furlong, E. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef]
- Pekowska, A.; Benoukraf, T.; Zacarias-Cabeza, J.; Belhocine, M.; Koch, F.; Holota, H.; Imbert, J.; Andrau, J.-C.; Ferrier, P.; Spicuglia, S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011, 30, 4198–4210. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Zhang, Y. Enhancer and super-enhancer: Positive regulators in gene transcription. Anim. Model Exp. Med. 2018, 1, 169–179. [Google Scholar] [CrossRef]
- Zhang, J.; Yue, W.; Zhou, Y.; Liao, M.; Chen, X.; Hua, J. Super enhancers-Functional cores under the 3D genome. Cell Prolif. 2021, 54, e12970. [Google Scholar] [CrossRef]
- Thomas, H.F.; Kotova, E.; Jayaram, S.; Pilz, A.; Romeike, M.; Lackner, A.; Penz, T.; Bock, C.; Leeb, M.; Halbritter, F.; et al. Temporal dissection of an enhancer cluster reveals distinct temporal and functional contributions of individual elements. Mol. Cell 2021, 81, 969–982. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, Y.-Y.; Lin, D.-C. Super-enhancer-mediated core regulatory circuitry in human cancer. Comput. Struct. Biotechnol. J. 2021, 19, 2790–2795. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Yang, Z.; Tan, Y.; Li, Y. Super-enhancer function and its application in cancer targeted therapy. NPJ Precis Oncol. 2020, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erokhin, M.; Vassetzky, Y.; Georgiev, P.; Chetverina, D. Eukaryotic enhancers: Common features, regulation, and participation in diseases. Cell. Mol. Life Sci. 2015, 72, 2361–2375. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hah, N.; Benner, C.; Chong, L.-W.; Yu, R.T.; Downes, M.; Evans, R.M. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc. Natl. Acad. Sci. USA 2015, 112, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of Cell Identity Genes Occurs in Insulated Neighborhoods in Mammalian Chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.; Rubin, A.; Flynn, R.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.; Wu, Q.; Liu, M.; Su, M.; Liu, S.; Shao, L.; Han, X.; He, H. EphA2 super-enhancer promotes tumor progression by recruiting FOSL2 and TCF7L2 to activate the target gene EphA2. Cell Death Dis. 2021, 12, 264. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef]
- Moffitt, J.; Zhuang, X. RNA Imaging with Multiplexed Error-Robust Fluorescence in Situ Hybridization (MERFISH). Methods Enzymol. 2016, 572, 1–49. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.W.; Hendrix, D.A.; Levine, M.S. Shadow enhancers as a source of evolutionary novelty. Science 2008, 321, 1314. [Google Scholar] [CrossRef] [Green Version]
- Osterwalder, M.; Barozzi, I.; Tissières, V.; Fukuda-Yuzawa, Y.; Mannion, B.J.; Afzal, S.Y.; Lee, E.A.; Zhu, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 2018, 554, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.X.; Xie, P.; Collings, C.K.; Cao, K.; Aoi, Y.; Marshall, S.A.; Rendleman, E.J.; Ugarenko, M.; Ozark, P.A.; Zhang, A.; et al. PAF1 regulation of promoter-proximal pause release via enhancer activation. Science 2017, 357, 1294–1298. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.X.; Woodfin, A.R.; Gardini, A.; Rickels, R.A.; Marshall, S.A.; Smith, E.R.; Shiekhattar, R.; Shilatifard, A. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 2015, 162, 1003–1015. [Google Scholar] [CrossRef] [Green Version]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, J.C. Back to basics:Sox genes. Dev. Dyn. 2007, 236, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Masui, S.; Nakatake, Y.; Toyooka, Y.; Shimosato, D.; Yagi, R.; Takahashi, K.; Okochi, H.; Okuda, A.; Matoba, R.; Sharov, A.A.; et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature 2007, 9, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Rodda, D.J.; Chew, J.-L.; Lim, L.-H.; Loh, Y.-H.; Wang, B.; Ng, H.-H.; Robson, P. Transcriptional Regulation of Nanog by OCT4 and SOX2. J. Biol. Chem. 2005, 280, 24731–24737. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Niec, R.E.; Kim, H.Y.; Treuting, P.; Chinen, T.; Zheng, Y.; Umetsu, D.T.; Rudensky, A.Y. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012, 482, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.J.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Feng, Y.; Van Der Veeken, J.; Shugay, M.; Putintseva, E.V.; Osmanbeyoglu, H.U.; Dikiy, S.; Hoyos, B.E.; Moltedo, B.; Hemmers, S.; Treuting, P.; et al. A mechanism for expansion of regulatory T-cell repertoire and its role in self-tolerance. Nature 2015, 528, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the Inheritance of Regulatory T Cell Identity by a cis Element in the Foxp3 Locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Capparelli, R.; Cuomo, P.; Gentile, A.; Iannelli, D. Microbiota-Liver Diseases Interactions. Int. J. Mol. Sci. 2023, 24, 3883. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Boissel, S.; Zarhrate, M.; Rio, M.; Munnich, A.; Egly, J.M.; Colleaux, L. MED23 mutation links intellectual disability to dysregulation of immediate early gene expression. Science 2011, 333, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- Trehan, A.; Brady, J.M.; Maduro, V.; Bone, W.P.; Huang, Y.; Golas, G.A.; Kane, M.S.; Lee, P.R.; Thurm, A.; Gropman, A.L.; et al. MED23-associated intellectual disability in a non-consanguineous family. Am. J. Med. Genet. A 2015, 167, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Hashemi-Gorji, F.; Fardaei, M.; Tabei, S.M.B.; Miryounesi, M. Novel mutation in the MED23 gene for intellectual disability: A case report and literature review. Clin. Case Rep. 2019, 7, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ohgi, K.A.; Rosenfeld, M.G.; Zhang, J.; Krones, A.; Bush, K.T.; Glass, C.K.; Nigam, S.K.; Aggarwal, A.K.; Maas, R.L.; et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003, 426, 247–254. [Google Scholar] [CrossRef]
- Barrington, C.; Georgopoulou, D.; Pezic, D.; Varsally, W.; Herrero, J.; Hadjur, S. Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology. Nat. Commun. 2019, 10, 2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muerdter, F.; Boryń, Ł.M.; Arnold, C.D. STARR-seq—Principles and applications. Genomics 2015, 106, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Chen, D.; Chen, Y. Advances of DNase-seq for mapping active gene regulatory elements across the genome in animals. Gene 2018, 667, 83–94. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.C.; Zhang, G.; Estève, P.-O.; Spracklin, G.; Sian, S.; Xu, S.-Y.; Benoukraf, T.; Pradhan, S. NicE-seq: High resolution open chromatin profiling. Genome Biol. 2017, 18, 122. [Google Scholar] [CrossRef] [Green Version]
- Snetkova, V.; Ypsilanti, A.R.; Akiyama, J.A.; Mannion, B.J.; Plajzer-Frick, I.; Novak, C.S.; Harrington, A.N.; Pham, Q.T.; Kato, M.; Zhu, Y.; et al. Ultraconserved enhancer function does not require perfect sequence conservation. Nat. Genet. 2021, 53, 521–528. [Google Scholar] [CrossRef]
- Villar, D.; Berthelot, C.; Aldridge, S.; Rayner, T.F.; Lukk, M.; Pignatelli, M.; Park, T.J.; Deaville, R.; Erichsen, J.T.; Jasinska, A.J.; et al. Enhancer Evolution across 20 Mammalian Species. Cell 2015, 160, 554–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.S.; Zheng, D.; Tan, S.Z.; Bower, N.I.; Garside, V.; Vanwalleghem, G.; Gaiti, F.; Scott, E.; Hogan, B.M.; Kikuchi, K.; et al. Deep conservation of the enhancer regulatory code in animals. Science 2020, 370, eaax8137. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Liu, Y.; Li, T.; Li, C. 3Disease Browser: A Web server for integrating 3D genome and disease-associated chromosome rearrangement data. Sci. Rep. 2016, 6, 34651. [Google Scholar] [CrossRef] [Green Version]
- Singer, J.; Irmisch, A.; Ruscheweyh, H.-J.; Singer, F.; Toussaint, N.C.; Levesque, M.P.; Stekhoven, D.J.; Beerenwinkel, N. Bioinformatics for precision oncology. Brief. Bioinform. 2019, 20, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Vitali, F.; Li, Q.; Schissler, A.G.; Berghout, J.; Kenost, C.; Lussier, Y.A. Developing a ‘personalome’ for precision medicine: Emerging methods that compute interpretable effect sizes from single-subject transcriptomes. Brief. Bioinform. 2019, 20, 789–805. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317. [Google Scholar] [CrossRef]
- Chang, Z.L.; Chen, Y.Y. CARs. synthetic immunoreceptors for cancer therapy and beyond. Trends Mol. Med. 2017, 23, 430–450. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Rajan, A.; Kennedy, J.J.; Ivey, R.G.; Shelby, S.A.; Leung, I.; Templeton, M.L.; Muhunthan, V.; Voillet, V.; Sommermeyer, D.; et al. Comparative Analysis of TCR and CAR Signaling Informs CAR Designs with Superior Antigen Sensitivity and In Vivo Function. Sci. Signal 2021, 14, eabe2606. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Tsumaki, N.; Okada, M.; Yamashita, A. iPS cell technologies and cartilage regeneration. Bone 2015, 70, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Gong, H.; Cui, L.; Zhang, W.; Ma, J.; Chen, C.; Ai, H.; Xiao, S.; Huang, L.; et al. Genetic correlation of fatty acid composition with growth, carcass, fat deposition and meat quality traits based on GWAS data in six pig populations. Meat Sci. 2019, 150, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Marina, H.; Pelayo, R.; Suárez-Vega, A.; Gutiérrez-Gil, B.; Esteban-Blanco, C.; Arranz, J. Genome-wide association studies (GWAS) and post-GWAS analyses for technological traits in Assaf and Churra dairy breeds. J. Dairy Sci. 2021, 104, 11850–11866. [Google Scholar] [CrossRef]
- Zhang, X.; Jamwal, K.; Distl, O. Tracking footprints of artificial and natural selection signatures in breeding and non-breeding cats. Sci. Rep. 2022, 12, 18061. [Google Scholar] [CrossRef]
- Chen, H.; Xie, W.; He, H.; Yu, H.; Chen, W.; Li, J.; Yu, R.; Yao, Y.; Zhang, W.; He, Y.; et al. A High-Density SNP Genotyping Array for Rice Biology and Molecular Breeding. Mol. Plant 2014, 7, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.Y.; Janss, L.L.; Andersen, J.R.; Orabi, J.; Jensen, J.D.; Jahoor, A.; Jensen, J. Genomic prediction and GWAS of yield, quality and disease-related traits in spring barley and winter wheat. Sci. Rep. 2020, 10, 3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Liu, D.; Luo, J.; Kong, D.; Zhang, Y. Exploring the Role of Enhancer-Mediated Transcriptional Regulation in Precision Biology. Int. J. Mol. Sci. 2023, 24, 10843. https://doi.org/10.3390/ijms241310843
Wang X, Liu D, Luo J, Kong D, Zhang Y. Exploring the Role of Enhancer-Mediated Transcriptional Regulation in Precision Biology. International Journal of Molecular Sciences. 2023; 24(13):10843. https://doi.org/10.3390/ijms241310843
Chicago/Turabian StyleWang, Xueyan, Danli Liu, Jing Luo, Dashuai Kong, and Yubo Zhang. 2023. "Exploring the Role of Enhancer-Mediated Transcriptional Regulation in Precision Biology" International Journal of Molecular Sciences 24, no. 13: 10843. https://doi.org/10.3390/ijms241310843
APA StyleWang, X., Liu, D., Luo, J., Kong, D., & Zhang, Y. (2023). Exploring the Role of Enhancer-Mediated Transcriptional Regulation in Precision Biology. International Journal of Molecular Sciences, 24(13), 10843. https://doi.org/10.3390/ijms241310843