1. Introduction
Hypertension is a major risk factor for many fatal cardiovascular complications, such as stroke and myocardial infarction, which are currently a leading cause of death in the US and worldwide [
1,
2]. Although conventional antihypertensive drugs have been successfully used in the clinic to control blood pressure, about 15% of hypertensive patients suffer from drug-resistant hypertension [
3]. Patients with resistant hypertension are at high risk of cardiovascular complications and mortality with few treatment options [
3]. Therefore, it is urgent to identify novel therapeutic targets in the cardiovascular system for treatment of hypertension.
Hypertension is characterized by enhanced vascular constriction and remodeling [
2]. Vascular remodeling, such as hypertrophy and stiffness, is a major pathological alteration associated with chronic hypertension, causing end-organ damage and leading to cardiovascular comorbidities [
4]. Vascular smooth muscle cells (VSMCs) play an important role in vascular remodeling, switching their phenotype from the differentiated contractile state to de-differentiated synthetic and proliferative states [
5]. Therefore, it is crucial to identify the regulatory mechanisms underlying VSMC phenotypic switching in order to prevent vascular remodeling, end-organ damage, and cardiovascular comorbidities in hypertension.
Large-conductance calcium-activated potassium (BK
Ca) channels are expressed in the plasma membrane (PM) of VSMCs and carry large-conductance outward potassium currents. The activity of BK
Ca is regulated by membrane potential and intracellular Ca
2+ concentration [
6]. Activation of this channel induces membrane potential hyperpolarization and voltage-sensitive L-type Ca
2+ channel inactivation in VSMCs, leading to vascular smooth muscle relaxation and vasodilation [
6]. Conversely, genetic deletion or inhibition of this channel causes membrane depolarization and increases in intracellular Ca
2+ concentration, which regulates contractile responses, gene expression, cell proliferation, and other cellular functions [
7,
8,
9]. Recent studies have demonstrated that reduced BK
Ca channel function in VSMCs is involved in the pathophysiology of hypertension, including increased arterial constriction and elevated cell-proliferation in response to vasoactive mediators, such as norepinephrine (NE) and angiotensin II (Ang II) [
7,
10]. Therefore, it has been proposed that reduced expression, post-translational modification, and membrane-trafficking of this channel protein may contribute to those vascular alterations in hypertension [
11,
12,
13]. However, the exact intracellular mechanisms involved in BK
Ca dysfunction in the pathogenesis of hypertension are not yet fully clear.
In this study, we investigated and compared the protein expression, functional alterations, and intracellular molecular regulation of the BKCa channel in hypertensive and normotensive animal models. Our goal was to identify the molecular mechanisms underlying BKCa channel alteration in VSMC phenotype switching and in the development of vascular remodeling during hypertension. By using multiple techniques, we found that the expression of the BKCa channel, as well as its Ca2+-sensitivity and voltage-dependency, were unchanged in VSMCs of hypertensive rats compared to normotensive rats. However, we did observe a reduction in the interaction between the BKCa channel in the plasma membrane (PM) and the inositol 1,4,5-trisphosphate receptor (IP3R) in the sarcoplasmic reticulum (SR) of VSMCs in hypertensive rats. We further investigated the role of this reduced interaction between IP3R-BKCa in VSMC proliferation and vascular hypertrophy in hypertension.
3. Discussion
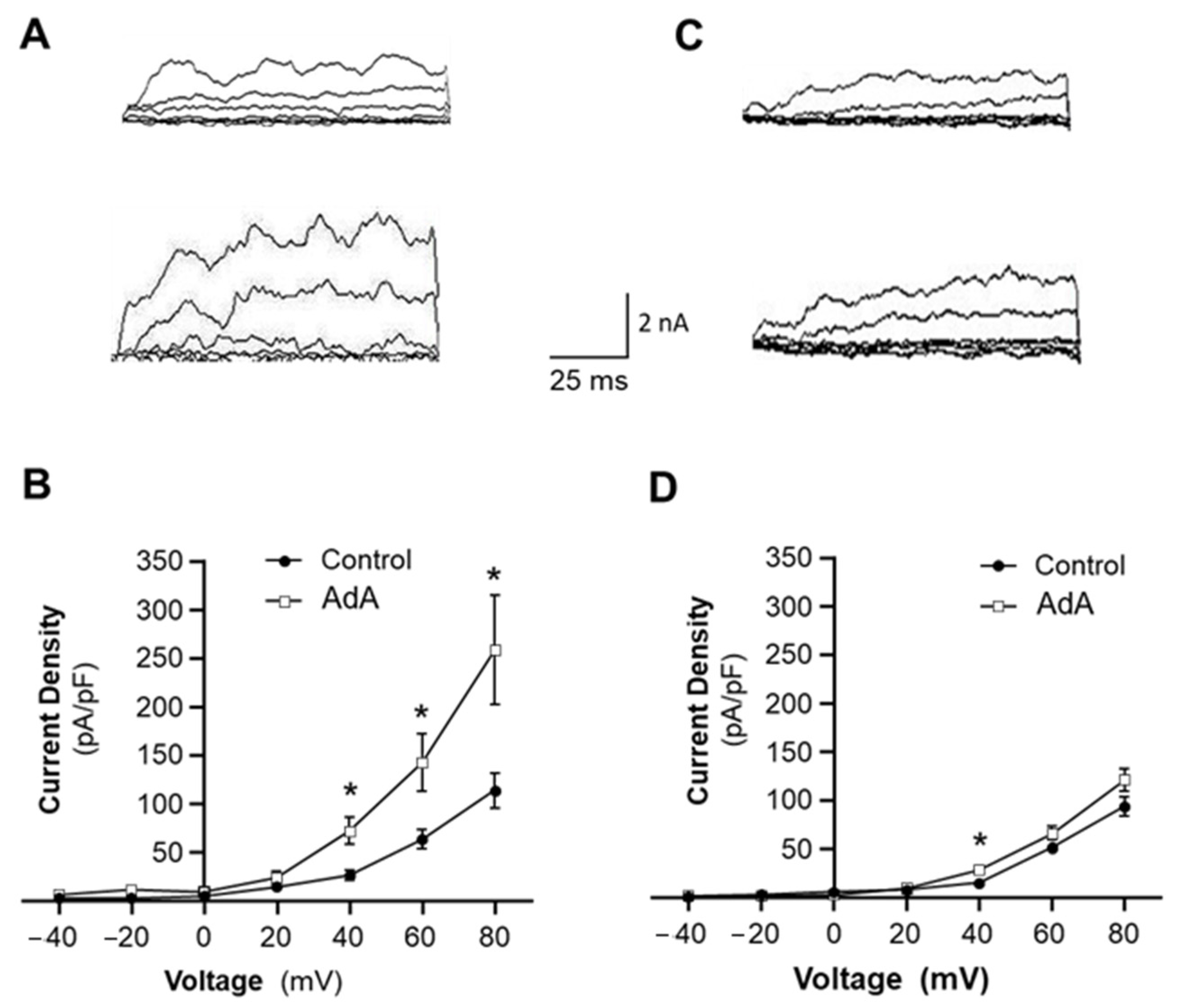
The present study provides the first evidence that disruption of IP3R-BKCa coupling in VSMCs is involved in vascular remodeling in hypertension. This conclusion is supported by the following evidence: (1) The Ca2+-sensitivity and voltage-dependence of BKCa channels are comparable in VSMCs from SHR and WKY rats. (2) an IP3R agonist increases BKCa channel currents, suggesting the functional coupling IP3R-BKCa, and the functional coupling of IP3R-BKCa is impaired in VSMCs of SHR. (3) Co-immunoprecipitation results indicate that IP3R-BKCa proteins are associated together, suggesting molecular coupling of IP3R-BKCa. The molecular coupling of IP3R-BKCa is reduced in mesenteric arteries of SHR. (4) Inhibition of BKCa channels augments Ang II-induced proliferation in VSMCs of WKY rats, mimicking the enhanced effect of Ang II in SHR. (5) Disrupting IP3-BKCa coupling using JPH2-shRNA also increases Ang II-induced VSMC proliferation in WKY rats. (6) Treatment with a BKCa channel opener, to functionally bypass IP3R-BKCa deficiency, attenuated the vascular hypertrophy in SHR. All together, these results indicate that loss of IP3R-BKCa coupling may contribute to vascular remodeling in hypertension. However, several concerns need to be addressed.
The first concern raised from this study is how do BK
Ca channels regulate the proliferation of VSMCs, leading to vascular remodeling in hypertension? The mechanism involved in BK
Ca-mediated VSMC proliferation is still not clear. One possibility could be mediated by the control of intracellular Ca
2+ concentration. It is well-known that BK
Ca channels control intracellular Ca
2+ by a negative feedback signaling pathway to prevent excessive elevation of intracellular Ca
2+ after vasoactive agonist stimulation [
8,
20]. BK
Ca dysfunction, such as loss of IP3R-BK
Ca coupling, should increase the Ca
2+ response to agonist stimuli, leading to intracellular Ca
2+ elevation. Intracellular Ca
2+ not only triggers vasocontraction, but also regulates proliferation-related gene expression in VSMCs [
8], thereby controlling the VSMC phenotype. It has been reported that intracellular Ca
2+ elevation induces NFAT (nuclear factor of activated T cells) dephosphorylation by activation of calcineurin, a Ca
2+-dependent phosphatase, in VSMCs [
20]. Dephosphorylated NFAT translocates into the nucleus, binding to several transcription coactivators, which in turn lead to VSMC proliferation and vascular remodeling [
8,
20]. This notion is supported by the current observation that BK
Ca channel inhibition or disruption of IP3R-BK
Ca coupling significantly increases Ang II-induced VSMC proliferation. This observation is also consistent with other observations showing that the disruption and blockade of BK
Ca function induces VSMC proliferation and vascular hypertrophy [
7,
10]. The results from this study add further supporting evidence by showing that treatment of SHR with a BK
Ca opener significantly attenuated the development of vascular remodeling.
The results from the current study show that BK
Ca and IP3R couple together to maintain BK
Ca channel activity. The next question raised from this study could be how is BK
Ca activity regulated by IP3R in VSMCs? IP3R is expressed in the SR, functioning as a Ca
2+ releasing channel from this intracellular Ca
2+ store [
14,
15,
16]. The IP3R is a receptor for inositol 1,4,5-trisphosphate (IP3), a plasma membrane lipid produced by phospholipase C (PLC) in response to the stimulation by vasoactive agonists, such as Ang II [
21]. The co-immunoprecipitation data in the current study demonstrate that BK
Ca and IP3R proteins are associated closely together. Considering BK
Ca channel activity is regulated by intracellular Ca
2+, it is possible that the IP3R-derived Ca
2+ release from the SR may stimulate BK
Ca activity. This hypothesis is supported by our whole-cell patch clamp studies showing that an IP3R agonist increases the BK
Ca current. In addition, results from other research groups also show the direct association of IP3R and BK
Ca channels in the PM-SR conjunction of VSMCs [
14]. However, the detailed molecular interactions in this signaling pathway in VSMCs in response to pathologic stimuli still need further investigation.
Interestingly, the coupling of IP3R-BK
Ca is impaired in VSMCs of SHR as compared with WKY rats. Considering that BK
Ca channel activation induced by IP3-induced Ca
2+ release provides an important negative feedback mechanism to protect against overresponse to vasoactive agonists [
22,
23], loss of coupling between IP3R-BK
Ca would induce BK
Ca dysfunction, thereby increasing the sensitivity of the blood vessels to agonist stimuli and leading to vascular hypertrophy and hyperplasia. This speculation is supported by data from our current study showing that blockade of BK
Ca channels or disruption of IP3R-BK
Ca coupling enhances Ang II-induced VSMC proliferation in WKY rats, mimicking the enhanced response to Ang II in SHR. Conversely, treatment with a BK
Ca channel opener, bypassing IP3R-BK
Ca decoupling, attenuates development of vascular hypertrophy in SHR. In this study, SHR, a genetic hypertension animal model, was used. Of course, all observations from SHR should be confirmed in other hypertensive animal models, as well as in vascular tissues of hypertensive humans in future studies.
While interest in studying the importance of SR-PM conjunction sites has grown tremendously recently, little is known about their role in the development of hypertension. The mechanism involved in IP3R-BK
Ca coupling during hypertension is still unknown. One possible mechanism of IP3R-BK
Ca decoupling in VSMCs of SHR could be mediated by a deficit of JPH2, the tether protein between SR-PM. This hypothesis is supported by our data showing that knockdown of JPH2 expression enhances Ang II-induced VSMC proliferation. This notion is also supported by previous investigations showing that JPH2-knockout mice develop severe cardiac hypertrophy [
24] and that JPH2 could be cleaved by calpain, an intracellular calcium-sensitive protease [
25]. Therefore, we can speculate that elevated calcium within VSMCs of SHR stimulates calpain, which cleaves JPH2, leading to IP3R-BK
Ca decoupling. However, this hypothesis requires further investigation and will be the focus of our future studies.
In conclusion, results from this study demonstrate that Ca2+-sensitivity, voltage-dependence, and protein expression of the BKCa channel in VSMCs are comparable between SHR and WKY rats. Impaired IP3R-BKCa coupling is observed in VSMCs of SHR as compared with WKY rats. This IP3R-BKCa decoupling in VSMCs of SHR may contribute to the vascular hypertrophy and remodeling observed in hypertension.
4. Materials and Methods
4.1. Animals and Materials
Experiments were performed on 4–6-month-old spontaneously hypertensive (SHR) rats and normotensive Wistar Kyoto (WKY) rats of either sex. The rats were purchased from Charles River (Wilmington, MA, USA) and housed under controlled conditions with a 12:12 h light–dark cycle. Food and water were available to the animals ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of North Dakota State University.
Paxilline, Angiotensin II (Ang II), and NS1619 were purchased from Cayman (Ann Arbor, ML, USA). Anti-SM actin antibody, Dulbecco’s modified Eagle medium (DMEM), and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Collagenase was purchased from Worthington Biochemical (Lakewood, NJ, USA). Anti IP3 receptor (IP3R)1 antibody and protein A/G plus agarose were obtained from Santa Cruz (Dallas, TX, USA). KCNMA1 antibody was purchased from Alomone Lab (Jerusalem, Israel). 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1, 3-benzene disulfonate (WST-1) was obtained from Abcam. 4′,6-diamidino-2-phenylindole (DAPI), Adenophostin A (AdA), Norepinephrine (NE), ATP, GTP, HEPES, and others were purchased from Sigma-Aldrich (St. Louis, MO, USA).
4.2. VSMC Isolation and Culture
The VSMCs were dissociated from small mesenteric arteries using enzymatic methods, as described in our previous publication [
26]. Briefly, small mesenteric arteries were dissected under a microscope at third- and fourth-order branches, then incubated for 10 min in 2 mL of low-Ca
2+ Tyrode’s solution containing (in mM) 145 NaCl, 4 KCl, 0.05 CaCl
2, 1 MgCl
2, 10 HEPES, and 10 dextrose, plus 1 mg/mL albumin, followed by 20 min at 37 °C in the same Tyrode’s solution containing 1.5 mg/mL papain and 1 mg/mL DTT. Finally, the arterial segments were incubated for 30 min at 37 °C in the Tyrode’s solution containing 2 mg/mL collagenase, 0.5 mg/mL elastase, and 1 mg/mL soybean trypsin inhibitor. Tissues were then triturated gently using a fire-polished wide-bore pipette to release single VSMCs. Cells were either stored in low-Ca
2+ Tyrode’s solution at 4 °C for electrophysiological experiments within 6 h or preparation of VSMC cultures.
4.3. VSMC Culture and Transfection
Dissociated mesenteric arterial VSMCs were cultured in 25 cm
2 culture flasks, which contained DMEM supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL). Cells were passaged as they became confluent, and cells at the 3rd–5th passages were used for experiments. The VSMC marker, α-smooth muscle actin (α-SMA), was used to identify the VSMCs by using immunocytochemistry, as described in our previous publication [
27]. The cultured VSMCs were used to study cell morphology and proliferation. To examine the role of IP3R-BK
Ca coupling in Ang II-induced alterations in cell morphology and proliferation, AAV2-mediated overexpression of Junctophilin-2 shRNA (AAV2-JPH2-shRNA) was used to knockdown Junctophilin-2 (JPH2) expression. JPH2 is the major tethering protein between the plasma membrane (PM) and sarcoplasmic reticulum (SR), facilitating IP3R-BK
Ca coupling [
19]. The AAV2-JPH2-shRNA and its scrambled control (AAV2-JPH2-SCR) were constructed and prepared as described in our previous publication [
28]. The AAV2-JPH2-shRNA or AAV2-JPH-SCR (all in 1 × 10
10 genome copies per microliter, 2 µL of AAV2 into 5 mL medium) were added to the culture medium. After three days of transduction, the cells were used for morphology and proliferation studies. The successful knockdown of JPH2 was verified using real-time PCR, as described in our previous publication [
29]. Briefly, TaqMan probe specific for rat JPH2 was purchased from Applied Biosystems Inc (Waltham, MA, USA). Real-time PCR was performed in an Applied Biosystem PRISM 700 sequence detection system, according to the protocol provided by the manufacturer. Data were normalized to 18S RNA. In each experiment, samples were analyzed in triplicate.
4.4. VSMC Proliferation Assay
Primarily cultured VSMCs from small mesenteric arteries of SHR and WKY rats were seeded onto glass coverslips in 35 mm dishes. After reaching 70% confluency, the cells were serum-starved for 24 h before treatment. Cells were stimulated with either the vehicle (PBS), Ang II (0.1 µM), or Ang II plus paxilline (a BKCa blocker, 1 μM) for 48 h. Cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 2% BSA for 1 h at room temperature. Cells were then stained with mouse anti-α-SMA antibody (1:250) in 0.1% BSA at 4 °C overnight, and then labeled with goat anti-mouse secondary antibody conjugated to Alexa Fluor 594 (1:500). The positive cell numbers were counted under a Carl Zeiss LSM 900 microscope (Carl Zeiss, Baden-Wurttemberg, Germany).
VSMC proliferation was detected using the WST-1 reagent. Cells were plated in a 96-well microplate and grown to 70% confluence. After being serum-starved for 24 h, cells were treated with vehicle (PBS), Ang II (0.1 µM), or Ang II plus paxilline (1 μM) for 24 h. The medium was then replaced with 100 µL of WST-1 (1:10 dilution) in fresh medium, followed by incubation for 3 h. Absorbance was measured using a multifunctional microplate reader (Spectra Max M5, Molecular Devices, San Jose, CA, USA) at 440 nm, with reference wavelength set at 630 nm.
4.5. Electrophysiological Recordings
BK
Ca channel activity in mesenteric VSMCs was recorded at room temperature, either in whole-cell configuration or from inside-out patches, as described in our previous publications [
26,
30]. An Axopatch 200B patch-clamp amplifier (Axon Instruments, Burlingame, CA, USA) and pCLAMP 10.0 software (Molecular Devices, San Jose, CA, USA) were used to control voltage-clamp and voltage-pulse generation. Voltage-activated currents were filtered at 1 kHz and digitized at 5 kHz. Leakage current was subtracted digitally. Series resistance and total cell capacitance were obtained by adjusting series resistance and whole-cell capacitance using the Axopatch 200B amplifier. For inside-out patches, the recording pipettes (resistance 5–6 MΩ) were filled with a solution containing (in mM) 145 KCl, 1.8 CaCl
2, MgCl
2 1.1, and 5 HEPES; pH 7.2 (KOH). Cells were bathed in a solution containing (in mM) 145 KCl, 1.1 MgCl
2, 0.37 CaCl
2, 10 HEPES, 1 EGTA, and 10 dextrose; pH 7.4 (KOH). Free Ca
2+ levels on the cytoplasmic face of the membrane were set by adding the calculated ratio of CaCl
2 and EGTA (using Chelator 1.0 software, Schoenmakers, Nijmen, The Netherlands). BK
Ca open-state probability (NPo) and unitary amplitudes of single-channel currents were obtained at different membrane potentials between −70 mV to +70 mV (20 mV steps) in the presence of 0.3, 1, 1.5, or 3 μM Ca
2+.
In whole-cell voltage-clamp experiments, the bath solution contained (in mM) 145 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, and 10 dextrose; pH 7.4 (NaOH). The recording pipettes (resistance 3–4 MΩ) were filled with a solution containing (in mM) 145 KCl, 5 NaCl, 0.37 CaCl2, 2 MgCl2, 10 HEPES, 1 EGTA, and 7.5 dextrose; pH 7.2 (KOH). Cells were held at −60 mV, and 100-millisecond depolarizing step pulses of 20 mV increments from –40 to +80 mV voltages were applied. The BKCa current was divided by the capacitance and expressed as current density. Analysis was performed offline using Clampfit 10 (Axon Instruments, Burlingame, CA, USA).
4.6. Western Blotting and Immunoprecipitation
BK
Ca and IP3R protein levels in rat mesenteric arteries were assessed by Western blot analysis, as in our previous publication [
27]. Briefly, proteins were separated on 7.5% polyacrylamide gels by SDS-PAGE and electroblotted onto a nitrocellulose membrane. Membranes were blocked in TBS-T (0.08% Tween) containing 5% milk for 1 h, followed by overnight incubation with rabbit polyclonal anti-KCNMA1 or rabbit polyclonal anti-IP3R1 primary antibodies at 4 °C. After washing with TBS-T, membranes were incubated for 1 h with anti-rabbit horseradish peroxidase–conjugated secondary antibodies. To ensure equal loading, the membranes were re-probed for β-actin after stripping. Membranes were developed using enhanced chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA), and digital images were obtained using an AGFA CP1000 automatic film processor. Relative protein expression values were obtained by dividing the raw values of BK
Ca and IP3R by the raw values of β-actin.
For co-immunoprecipitation, mesenteric arteries were lysed in non-denaturizing cell lysis buffer (Abcam, Cambridge, UK) containing a protease inhibitor mixture (Thermo Fisher Scientific, Waltham, MA, USA). The cell lysate (1.5 mg) was incubated with 8 μg rabbit polyclonal anti-KCNMA1 antibody for 2 h, followed by addition of 20 μL protein A/G PLUS–agarose beads (Santa Cruz Biotechnology, Dallas, TX, USA) for 12 h at 4 °C. After the incubation, samples were spun down and washed three times with PBS. Protein contents were then eluted with 2× SDS sample buffer. Total cell lysate was used as the positive control, while empty beads combined with cell lysate without anti-KCNMA1 antibody was used as the negative control. Samples were analyzed using conventional Western blots with mouse monoclonal anti-IP3R1 primary antibody and horseradish peroxidase–conjugated anti-mouse secondary antibody.
4.7. Vascular Morphology Detection
SHR and WKY rats were used to examine the effect of a BK
Ca channel opener on vascular remodeling. The BK
Ca channel opener, NS1619 (20 µg/kg/day), was administered chronically by subcutaneous infusion via osmotic minipumps (ALZET, Model 2004). The minipump implantation was performed as described in our previous publication [
27]. After chronic treatment with vehicle control or NS1619 (20 µg/kg/day) for 4 weeks, SHR and WKY rats were euthanized with overdoses of pentobarbital. The mesentery was isolated and cross-sectioned using a Cryostat (Leica Biosystems). The mesenteric artery sections were fixed in 4% formalin/PBS, followed by staining with hematoxylin and eosin (H&E), as described in our previous publication [
27]. The vascular morphology was visualized by a microscope (Olympus, Shinjuku City, Japan). Vascular wall thickness was measured with Infinity Capture and Analysis Software (v6.5.6) to evaluate vascular hypertrophy. The external diameter of the arteries measured was 100–150 µm in all arterial cross-sections.
4.8. Data Analysis
Results are expressed as means ± SE. Statistical significance was evaluated by one- or two-way ANOVA, as appropriate, followed by either Newman–Keuls or Bonferroni post hoc analysis, where appropriate. Differences were considered significant at p < 0.05. Individual probability values are noted in the figure legends.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






