
Experimental Models of Traumatic Injuries: Do They Capture the Coagulopathy and Underlying Endotheliopathy Induced by Human Trauma?
Abstract
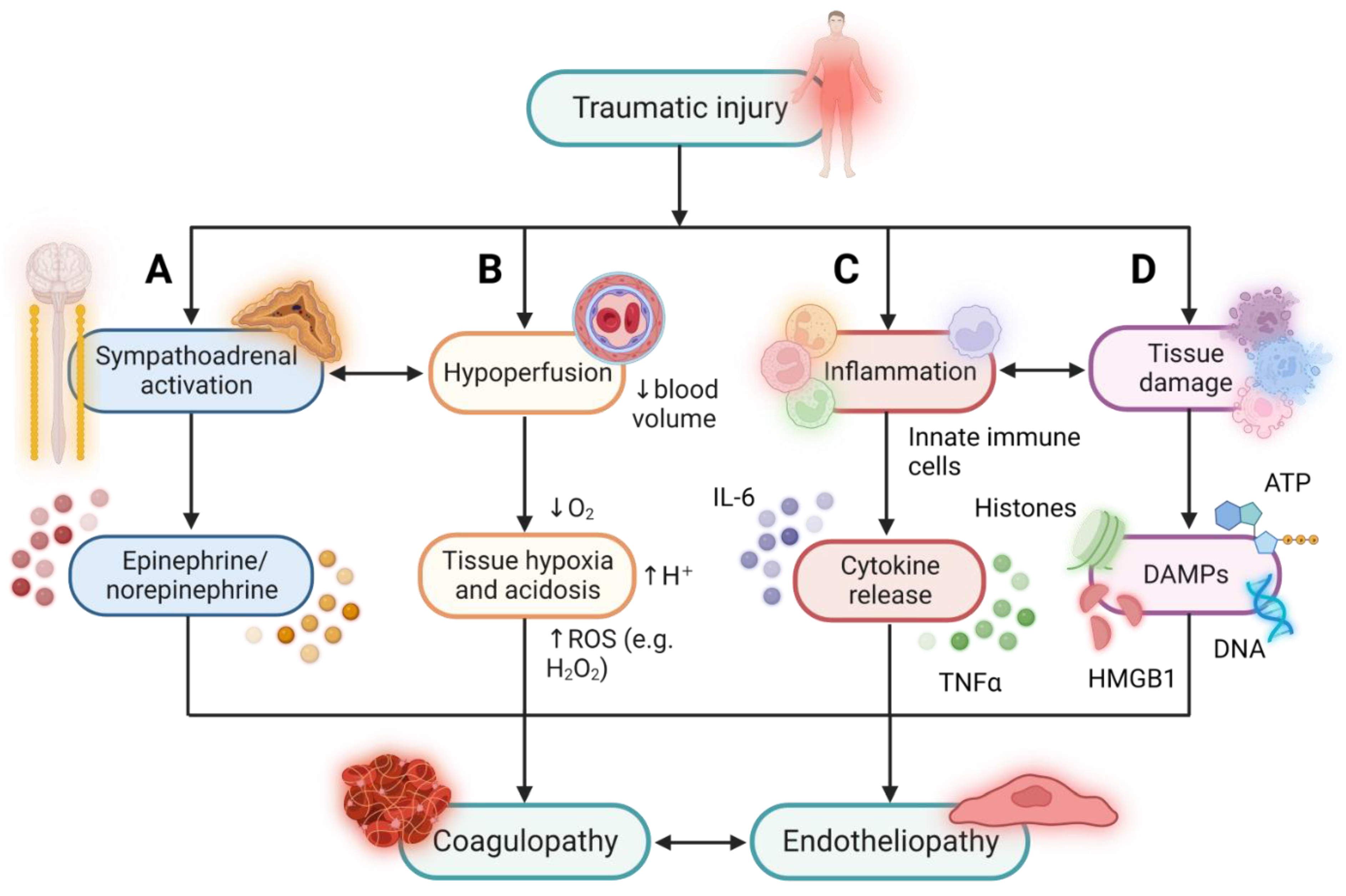
:1. Background
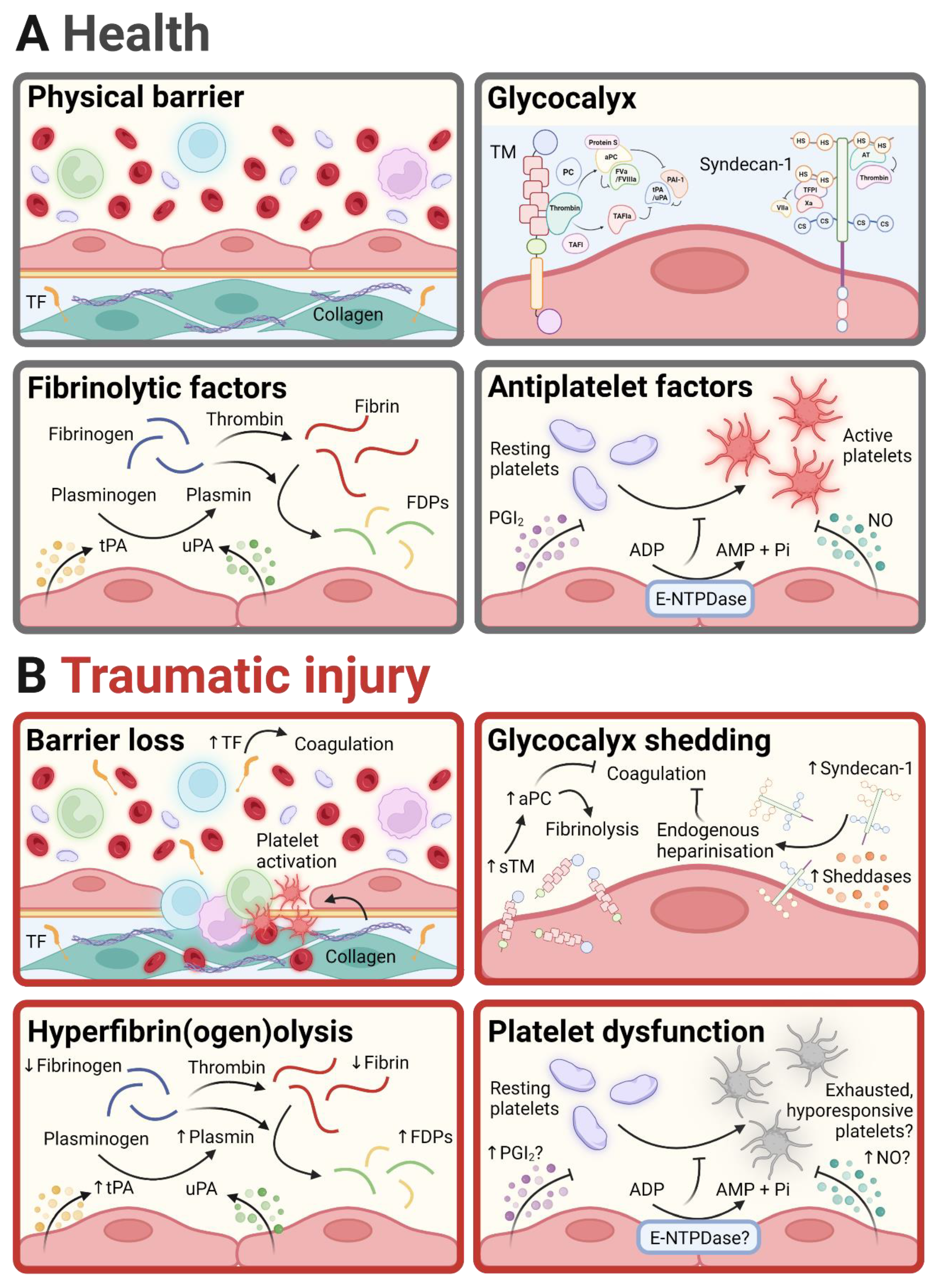
2. Endothelial Regulation of Haemostasis in Health and Following Traumatic Injury
3. In Vitro Models of Traumatic Injury
4. In Vivo Models of Traumatic Injury
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rossiter, N.D. Trauma-the forgotten pandemic? Int. Orthop. 2022, 46, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Collaborators GMaCoD. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clare, D.; Zink, K.L. Geriatric Trauma. Emerg. Med. Clin. N. Am. 2021, 39, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Atinga, A.; Shekkeris, A.; Fertleman, M.; Batrick, N.; Kashef, E.; Dick, E. Trauma in the elderly patient. Br. J. Radiol. 2018, 91, 20170739. [Google Scholar] [CrossRef]
- Curry, N.S.; Davenport, R.A.; Hunt, B.J.; Stanworth, S.J. Transfusion strategies for traumatic coagulopathy. Blood Rev. 2012, 26, 223–232. [Google Scholar] [CrossRef]
- Davenport, R.; Khan, S. Management of major trauma haemorrhage: Treatment priorities and controversies. Br. J. Haematol. 2011, 155, 537–548. [Google Scholar] [CrossRef]
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Duranteau, J.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Maegele, M.; Nardi, G.; Riddez, L.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef] [Green Version]
- Moran, C.G.; Lecky, F.; Bouamra, O.; Lawrence, T.; Edwards, A.; Woodford, M.; Willett, K.; Coats, T.J. Changing the System—Major Trauma Patients and Their Outcomes in the NHS (England) 2008–17. eClinicalMedicine 2018, 2–3, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Stanworth, S.J.; Davenport, R.; Curry, N.; Seeney, F.; Eaglestone, S.; Edwards, A.; Martin, K.; Allard, S.; Woodford, M.; Lecky, F.E.; et al. Mortality from trauma haemorrhage and opportunities for improvement in transfusion practice. Br. J. Surg. 2016, 103, 357–365. [Google Scholar] [CrossRef]
- David, S.D.; Aroke, A.; Roy, N.; Solomon, H.; Lundborg, C.S.; Gerdin Wärnberg, M. Measuring socioeconomic outcomes in trauma patients up to one year post-discharge: A systematic review and meta-analysis. Injury 2022, 53, 272–285. [Google Scholar] [CrossRef]
- Moore, E.E.; Moore, H.B.; Kornblith, L.Z.; Neal, M.D.; Hoffman, M.; Mutch, N.J.; Schöchl, H.; Hunt, B.J.; Sauaia, A. Trauma-induced coagulopathy. Nat. Rev. Dis. Primers 2021, 7, 30. [Google Scholar] [CrossRef]
- Petros, S. Trauma-Induced Coagulopathy. Hamostaseologie 2019, 39, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornblith, L.Z.; Moore, H.B.; Cohen, M.J. Trauma-induced coagulopathy: The past, present, and future. J. Thromb. Haemost. 2019, 17, 852–862. [Google Scholar] [CrossRef]
- Duque, P.; Calvo, A.; Lockie, C.; Schöchl, H. Pathophysiology of Trauma-Induced Coagulopathy. Transfus. Med. Rev. 2021, 35, 80–86. [Google Scholar] [CrossRef]
- Ask, A.; Eltringham-Smith, L.; Bhakta, V.; Donkor, D.A.; Pryzdial, E.L.G.; Sheffield, W.P. Spotlight on animal models of acute traumatic coagulopathy: An update. Transfus. Apher. Sci. 2022, 61, 103412. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Caneva, L.; Gerosa, S.; Ricevuti, G. Trauma-Induced Coagulopathy: Overview of an Emerging Medical Problem from Pathophysiology to Outcomes. Medicines 2021, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M. Pathophysiology of trauma-induced coagulopathy: Disseminated intravascular coagulation with the fibrinolytic phenotype. J. Intensive Care 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Brøchner, A.C.; Toft, P. Pathophysiology of the systemic inflammatory response after major accidental trauma. Scand. J. Trauma Resusc. Emerg. Med. 2009, 17, 43. [Google Scholar] [CrossRef] [Green Version]
- Pape, H.C.; Moore, E.E.; McKinley, T.; Sauaia, A. Pathophysiology in patients with polytrauma. Injury 2022, 53, 2400–2412. [Google Scholar] [CrossRef]
- Dufour-Gaume, F.; Frescaline, N.; Cardona, V.; Prat, N.J. Danger signals in traumatic hemorrhagic shock and new lines for clinical applications. Front. Physiol. 2022, 13, 999011. [Google Scholar] [CrossRef] [PubMed]
- Peltz, E.D.; Moore, E.E.; Eckels, P.C.; Damle, S.S.; Tsuruta, Y.; Johnson, J.L.; Sauaia, A.; Silliman, C.C.; Banerjee, A.; Abraham, E. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock 2009, 32, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gögenur, M.; Burcharth, J.; Gögenur, I. The role of total cell-free DNA in predicting outcomes among trauma patients in the intensive care unit: A systematic review. Crit. Care 2017, 21, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Trulson, I.; Stahl, J.; Margraf, S.; Scholz, M.; Hoecherl, E.; Wolf, K.; Durner, J.; Klawonn, F.; Holdenrieder, S. Cell-Free DNA in Plasma and Serum Indicates Disease Severity and Prognosis in Blunt Trauma Patients. Diagnostics 2023, 13, 1150. [Google Scholar] [CrossRef]
- Binkowska, A.M.; Michalak, G.; Pilip, S.; Kopacz, M.; Słotwiński, R. The diagnostic value of early cytokine response in patients after major trauma—Preliminary report. Cent. Eur. J. Immunol. 2018, 43, 33–41. [Google Scholar] [CrossRef]
- Hildebrand, F.; Pape, H.C.; Krettek, C. The importance of cytokines in the posttraumatic inflammatory reaction. Unfallchirurg 2005, 108, 793–794, 796–803. [Google Scholar] [CrossRef]
- Johansson, P.I.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Ostrowski, S.R. Traumatic Endotheliopathy: A Prospective Observational Study of 424 Severely Injured Patients. Ann. Surg. 2017, 265, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Krocker, J.D.; Lee, K.H.; Henriksen, H.H.; Wang, Y.W.; Schoof, E.M.; Karvelsson, S.T.; Rolfsson, Ó.; Johansson, P.I.; Pedroza, C.; Wade, C.E. Exploratory Investigation of the Plasma Proteome Associated with the Endotheliopathy of Trauma. Int. J. Mol. Sci. 2022, 23, 6123. [Google Scholar] [CrossRef]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L.; Committee, E.C. Thrombotic Regulation From the Endothelial Cell Perspectives. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e90–e95. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Endothelium and haemostasis. Hamostaseologie 2015, 35, 11–16. [Google Scholar] [CrossRef]
- Yang, Z.; Le, T.D.; Simovic, M.O.; Liu, B.; Fraker, T.L.; Cancio, T.S.; Cap, A.P.; Wade, C.E.; DalleLucca, J.J.; Li, Y. Traumatized triad of complementopathy, endotheliopathy, and coagulopathy—Impact on clinical outcomes in severe polytrauma patients. Front. Immunol. 2022, 13, 991048. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerome, W.G.; Handt, S.; Hantgan, R.R. Endothelial cells organize fibrin clots into structures that are more resistant to lysis. Microsc. Microanal. 2005, 11, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial Barrier and Its Abnormalities in Cardiovascular Disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, G.; Cirillo, P. Tissue factor: Newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc. Diagn. Ther. 2018, 8, 581–593. [Google Scholar] [CrossRef]
- Rao, L.V.; Kothari, H.; Pendurthi, U.R. Tissue factor encryption and decryption: Facts and controversies. Thromb. Res. 2012, 129 (Suppl. S2), S13–S17. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Nuyttens, B.P.; Thijs, T.; Deckmyn, H.; Broos, K. Platelet adhesion to collagen. Thromb. Res. 2011, 127 (Suppl. S2), S26–S29. [Google Scholar] [CrossRef]
- Patterson, E.K.; Cepinskas, G.; Fraser, D.D. Endothelial Glycocalyx Degradation in Critical Illness and Injury. Front. Med. 2022, 9, 898592. [Google Scholar] [CrossRef] [PubMed]
- Villalba, N.; Baby, S.; Yuan, S.Y. The Endothelial Glycocalyx as a Double-Edged Sword in Microvascular Homeostasis and Pathogenesis. Front. Cell Dev. Biol. 2021, 9, 711003. [Google Scholar] [CrossRef]
- Moore, K.H.; Murphy, H.A.; George, E.M. The glycocalyx: A central regulator of vascular function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R508–R518. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kostidis, S.; Tiemeier, G.L.; Sol, W.M.P.J.; de Vries, M.R.; Giera, M.; Carmeliet, P.; van den Berg, B.M.; Rabelink, T.J. Shear Stress Regulation of Endothelial Glycocalyx Structure Is Determined by Glucobiosynthesis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 350–364. [Google Scholar] [CrossRef]
- Mast, A.E. Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Rodriguez, E.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S.; Henriksen, H.H.; Stensballe, J.; Cotton, B.A.; Holcomb, J.B.; Johansson, P.I.; et al. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, S.R.; Johansson, P.I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 73, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.H.; Aquino, R.S.; Park, P.W. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012, 31, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Floer, M.; Clausen, M.; Meister, T.; Vollenberg, R.; Bettenworth, D.; Tepasse, P.R. Soluble syndecan-1 as marker of intestinal inflammation: A preliminary study and evaluation of a new panel of biomarkers for non-invasive prediction of active ulcerative colitis. Adv. Clin. Exp. Med. 2021, 30, 655–660. [Google Scholar] [CrossRef]
- Couchman, J.R. Syndecans: Proteoglycan regulators of cell-surface microdomains? Nat. Rev. Mol. Cell Biol. 2003, 4, 926–937. [Google Scholar] [CrossRef]
- Boron, M.; Hauzer-Martin, T.; Keil, J.; Sun, X.L. Circulating Thrombomodulin: Release Mechanisms, Measurements, and Levels in Diseases and Medical Procedures. TH Open 2022, 6, e194–e212. [Google Scholar] [CrossRef]
- Watanabe-Kusunoki, K.; Nakazawa, D.; Ishizu, A.; Atsumi, T. Thrombomodulin as a Physiological Modulator of Intravascular Injury. Front. Immunol. 2020, 11, 575890. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Thachil, J.; Asakura, H.; Levy, J.H.; Iba, T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions-a multi-faceted anticoagulant protein with therapeutic potential. Crit. Care 2019, 23, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plug, T.; Meijers, J.C. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J. Thromb. Haemost. 2016, 14, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouma, B.N.; Meijers, J.C. Thrombin-activatable fibrinolysis inhibitor (TAFI, plasma procarboxypeptidase B, procarboxypeptidase R, procarboxypeptidase U). J. Thromb. Haemost. 2003, 1, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Matthay, M.A.; Mackersie, R.C.; Pittet, J.F. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann. Surg. 2007, 245, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Davenport, R.A.; Guerreiro, M.; Frith, D.; Rourke, C.; Platton, S.; Cohen, M.; Pearse, R.; Thiemermann, C.; Brohi, K. Activated Protein C Drives the Hyperfibrinolysis of Acute Traumatic Coagulopathy. Anesthesiology 2017, 126, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Kutcher, M.E.; Xu, J.; Vilardi, R.F.; Ho, C.; Esmon, C.T.; Cohen, M.J. Extracellular histone release in response to traumatic injury: Implications for a compensatory role of activated protein C. J. Trauma Acute Care Surg. 2012, 73, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.J.; Call, M.; Nelson, M.; Calfee, C.S.; Esmon, C.T.; Brohi, K.; Pittet, J.F. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann. Surg. 2012, 255, 379–385. [Google Scholar] [CrossRef] [Green Version]
- McCachren, S.S.; Diggs, J.; Weinberg, J.B.; Dittman, W.A. Thrombomodulin expression by human blood monocytes and by human synovial tissue lining macrophages. Blood 1991, 78, 3128–3132. [Google Scholar] [CrossRef] [Green Version]
- Satta, N.; Freyssinet, J.M.; Toti, F. The significance of human monocyte thrombomodulin during membrane vesiculation and after stimulation by lipopolysaccharide. Br. J. Haematol. 1997, 96, 534–542. [Google Scholar] [CrossRef]
- Knipe, L.; Meli, A.; Hewlett, L.; Bierings, R.; Dempster, J.; Skehel, P.; Hannah, M.J.; Carter, T. A revised model for the secretion of tPA and cytokines from cultured endothelial cells. Blood 2010, 116, 2183–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood, E.C.; Hajjar, K.A. The annexin A2 system and vascular homeostasis. Vasc. Pharmacol. 2011, 54, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Choi, K.S.; Kassam, G.; Fitzpatrick, S.L.; Kwon, M.; Waisman, D.M. Role of annexin II tetramer in plasminogen activation. Trends Cardiovasc. Med. 1999, 9, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci. 2005, 10, 300–325. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Myrvang, H.K.; Dekker, L.V. Annexin A2 complexes with S100 proteins: Structure, function and pharmacological manipulation. Br. J. Pharmacol. 2015, 172, 1664–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madureira, P.A.; Surette, A.P.; Phipps, K.D.; Taboski, M.A.; Miller, V.A.; Waisman, D.M. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood 2011, 118, 4789–4797. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, M. Dynamics of fibrinogen in acute phases of trauma. J. Intensive Care 2017, 5, 3. [Google Scholar] [CrossRef]
- Schlimp, C.J.; Ponschab, M.; Voelckel, W.; Treichl, B.; Maegele, M.; Schöchl, H. Fibrinogen levels in trauma patients during the first seven days after fibrinogen concentrate therapy: A retrospective study. Scand. J. Trauma Resusc. Emerg. Med. 2016, 24, 29. [Google Scholar] [CrossRef] [Green Version]
- Puy, C.; Ngo, A.T.P.; Pang, J.; Keshari, R.S.; Hagen, M.W.; Hinds, M.T.; Gailani, D.; Gruber, A.; Lupu, F.; McCarty, O.J.T. Endothelial PAI-1 (Plasminogen Activator Inhibitor-1) Blocks the Intrinsic Pathway of Coagulation, Inducing the Clearance and Degradation of FXIa (Activated Factor XI). Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1390–1401. [Google Scholar] [CrossRef]
- Handt, S.; Jerome, W.G.; Tietze, L.; Hantgan, R.R. Plasminogen activator inhibitor-1 secretion of endothelial cells increases fibrinolytic resistance of an in vitro fibrin clot: Evidence for a key role of endothelial cells in thrombolytic resistance. Blood 1996, 87, 4204–4213. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.P.; Moore, E.E.; Moore, H.B.; Gonzalez, E.; Gamboni, F.; Chandler, J.G.; Mitra, S.; Ghasabyan, A.; Chin, T.L.; Sauaia, A.; et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg. 2016, 80, 16–23; discussion 23–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, J.C.; Matijevic, N.; Baer, L.A.; Holcomb, J.B.; Cotton, B.A.; Wade, C.E. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock 2014, 41, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Gall, L.S.; Vulliamy, P.; Gillespie, S.; Jones, T.F.; Pierre, R.S.J.; Breukers, S.E.; Gaarder, C.; Juffermans, N.P.; Maegele, M.; Stensballe, J.; et al. The S100A10 Pathway Mediates an Occult Hyperfibrinolytic Subtype in Trauma Patients. Ann. Surg. 2019, 269, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. The anti-aggregating properties of vascular endothelium: Interactions between prostacyclin and nitric oxide. Br. J. Pharmacol. 1987, 92, 639–646. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, T.M.; Zimmerman, G.A.; Satoh, K.; Prescott, S.M. Cultured endothelial cells synthesize both platelet-activating factor and prostacyclin in response to histamine, bradykinin, and adenosine triphosphate. J. Clin. Investig. 1985, 76, 271–280. [Google Scholar] [CrossRef]
- Sato, T.; Sawada, S.; Tsuda, Y.; Komatsu, S.; Akamatsu, N.; Kono, Y.; Higaki, T.; Imamura, H.; Tada, Y.; Yamasaki, S.; et al. The mechanism of thrombin-induced prostacyclin synthesis in human endothelial cells with reference to the gene transcription of prostacyclin-related enzymes and Ca2+ kinetics. J. Pharmacol. Toxicol. Methods 1999, 41, 173–182. [Google Scholar] [CrossRef]
- Gkaliagkousi, E.; Ritter, J.; Ferro, A. Platelet-derived nitric oxide signaling and regulation. Circ. Res. 2007, 101, 654–662. [Google Scholar] [CrossRef]
- Figueroa, X.F.; Poblete, I.; Fernández, R.; Pedemonte, C.; Cortés, V.; Huidobro-Toro, J.P. NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H134–H143. [Google Scholar] [CrossRef]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 2011, 61, 301–332. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, T.A.; Goswami, J.; Moon Tasson, L.; Tischer, A.; Bailey, K.R.; Spears, G.M.; Dong, J.F.; Auton, M.; Kozar, R.; Park, M.S. Quantification of von Willebrand factor and ADAMTS-13 after traumatic injury: A pilot study. Trauma Surg. Acute Care Open 2021, 6, e000703. [Google Scholar] [CrossRef]
- Matsumoto, H.; Takeba, J.; Umakoshi, K.; Kikuchi, S.; Ohshita, M.; Annen, S.; Moriyama, N.; Nakabayashi, Y.; Sato, N.; Aibiki, M. ADAMTS13 activity decreases in the early phase of trauma associated with coagulopathy and systemic inflammation: A prospective observational study. Thromb. J. 2021, 19, 17. [Google Scholar] [CrossRef]
- Martin, J.V.; Liberati, D.M.; Diebel, L.N. Disparate effects of catecholamines under stress conditions on endothelial glycocalyx injury: An in vitro model. Am. J. Surg. 2017, 214, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Diebel, L.N.; Martin, J.V.; Liberati, D.M. Microfluidics: A high-throughput system for the assessment of the endotheliopathy of trauma and the effect of timing of plasma administration on ameliorating shock-associated endothelial dysfunction. J. Trauma Acute Care Surg. 2018, 84, 575–582. [Google Scholar] [CrossRef]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Randi, A.M. Recent Advances in Endothelial Colony Forming Cells Toward Their Use in Clinical Translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.D.; Paschalaki, K.E.; Dyer, C.E.; Harrison-Lavoie, K.J.; Cutler, J.A.; McKinnon, T.A.; Millar, C.M.; Cutler, D.F.; Laffan, M.A.; Randi, A.M. Cellular and molecular basis of von Willebrand disease: Studies on blood outgrowth endothelial cells. Blood 2013, 121, 2773–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smadja, D.M.; Gaussem, P.; Mauge, L.; Israël-Biet, D.; Dignat-George, F.; Peyrard, S.; Agnoletti, G.; Vouhé, P.R.; Bonnet, D.; Lévy, M. Circulating endothelial cells: A new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation 2009, 119, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Sabulski, A.; Abdullah, S.; Luebbering, N.; Aunins, B.; Castillo, C.; Lake, K.; Duell, A.; Strecker, L.; Giordullo, L.; Broomhead, W.; et al. Circulating endothelial cells and the study of vascular injury in children undergoing hematopoietic stem cell transplant. Haematologica 2022, 107, 2950–2954. [Google Scholar] [CrossRef]
- Sakurai, Y.; Hardy, E.T.; Ahn, B.; Tran, R.; Fay, M.E.; Ciciliano, J.C.; Mannino, R.G.; Myers, D.R.; Qiu, Y.; Carden, M.A.; et al. A microengineered vascularized bleeding model that integrates the principal components of hemostasis. Nat. Commun. 2018, 9, 509. [Google Scholar] [CrossRef] [Green Version]
- Poventud-Fuentes, I.; Kwon, K.W.; Seo, J.; Tomaiuolo, M.; Stalker, T.J.; Brass, L.F.; Huh, D. A Human Vascular Injury-on-a-Chip Model of Hemostasis. Small 2021, 17, e2004889. [Google Scholar] [CrossRef] [PubMed]
- Shirure, V.S.; Hughes, C.C.W.; George, S.C. Engineering Vascularized Organoid-on-a-Chip Models. Annu. Rev. Biomed. Eng. 2021, 23, 141–167. [Google Scholar] [CrossRef]
- Frith, D.; Cohen, M.J.; Brohi, K. Animal models of trauma-induced coagulopathy. Thromb. Res. 2012, 129, 551–556. [Google Scholar] [CrossRef]
- van Zyl, N.; Reade, M.C.; Fraser, J.F. Experimental Animal Models of Traumatic Coagulopathy: A Systematic Review. Shock 2015, 44, 16–24. [Google Scholar] [CrossRef]
- Breschi, A.; Gingeras, T.R.; Guigó, R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 2017, 18, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Miyakawa, T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Martini, J.; Cabrales, P.; Fries, D.; Intaglietta, M.; Tsai, A.G. Effects of fibrinogen concentrate after shock/resuscitation: A comparison between in vivo microvascular clot formation and thromboelastometry. Crit. Care Med. 2013, 41, e301–e308. [Google Scholar] [CrossRef] [Green Version]
- Gangloff, C.; Grimault, O.; Theron, M.; Pichavant, K.; Galinat, H.; Mingant, F.; Ozier, Y. A clinically relevant and bias-controlled murine model to study acute traumatic coagulopathy. Sci. Rep. 2018, 8, 5783. [Google Scholar] [CrossRef] [Green Version]
- Chipman, A.M.; Wu, F.; Kozar, R.A. Fibrinogen inhibits microRNA-19b, a novel mechanism for repair of haemorrhagic shock-induced endothelial cell dysfunction. Blood Transfus. 2021, 19, 420–427. [Google Scholar] [CrossRef]
- Wu, F.; Wang, J.Y.; Chao, W.; Sims, C.; Kozar, R.A. miR-19b targets pulmonary endothelial syndecan-1 following hemorrhagic shock. Sci. Rep. 2020, 10, 15811. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.E.; Prakash, V.S.; Vercammen, J.M.; Pritts, T.; Kibbe, M.R. Development and validation of 4 different rat models of uncontrolled hemorrhage. JAMA Surg. 2015, 150, 316–324. [Google Scholar] [CrossRef]
- Hayakawa, M.; Tsuchida, T.; Honma, Y.; Mizugaki, A.; Ooyasu, T.; Yoshida, T.; Saito, T.; Katabami, K.; Wada, T.; Maekawa, K. Fibrinolytic system activation immediately following trauma was quickly and intensely suppressed in a rat model of severe blunt trauma. Sci. Rep. 2021, 11, 20283. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Gando, S.; Ono, Y.; Wada, T.; Yanagida, Y.; Sawamura, A.; Ieko, M. Noble-Collip Drum Trauma Induces Disseminated Intravascular Coagulation but Not Acute Coagulopathy of Trauma-Shock. Shock 2015, 43, 261–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesebro, B.B.; Rahn, P.; Carles, M.; Esmon, C.T.; Xu, J.; Brohi, K.; Frith, D.; Pittet, J.F.; Cohen, M.J. Increase in activated protein C mediates acute traumatic coagulopathy in mice. Shock 2009, 32, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, M.; Trivedi, A.; Miyazawa, B.Y.; Vivona, L.R.; Khakoo, M.; Zhang, H.; Pathipati, P.; Bagri, A.; Gatmaitan, M.G.; Kozar, R.; et al. Cryoprecipitate attenuates the endotheliopathy of trauma in mice subjected to hemorrhagic shock and trauma. J. Trauma Acute Care Surg. 2021, 90, 1022–1031. [Google Scholar] [CrossRef]
- White, N.J.; Martin, E.J.; Brophy, D.F.; Ward, K.R. Coagulopathy and traumatic shock: Characterizing hemostatic function during the critical period prior to fluid resuscitation. Resuscitation 2010, 81, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yu, W.K.; Lin, Z.L.; Tan, S.J.; Bai, X.W.; Ding, K.; Li, N. Chemical sympathectomy attenuates inflammation, glycocalyx shedding and coagulation disorders in rats with acute traumatic coagulopathy. Blood Coagul. Fibrinolysis 2015, 26, 152–160. [Google Scholar] [CrossRef]
- Wallen, T.E.; Singer, K.E.; Elson, N.C.; Baucom, M.R.; England, L.G.; Schuster, R.M.; Pritts, T.A.; Goodman, M.D. Defining Endotheliopathy in Murine Polytrauma Models. Shock 2022, 57, 291–298. [Google Scholar] [CrossRef]
- Darlington, D.N.; Craig, T.; Gonzales, M.D.; Schwacha, M.G.; Cap, A.P.; Dubick, M.A. Acute coagulopathy of trauma in the rat. Shock 2013, 39, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Darlington, D.N.; Cap, A.P. Procoagulant and fibrinolytic activity after polytrauma in rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R323–R329. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhao, K.; Shen, X.; Fan, X.X.; Ding, K.; Liu, R.M.; Wang, F. Blockade of Extracellular High-Mobility Group Box 1 Attenuates Systemic Inflammation and Coagulation Abnormalities in Rats with Acute Traumatic Coagulopathy. Med. Sci. Monit. 2016, 22, 2561–2570. [Google Scholar] [CrossRef] [Green Version]
- Frith, D.; Goslings, J.C.; Gaarder, C.; Maegele, M.; Cohen, M.J.; Allard, S.; Johansson, P.I.; Stanworth, S.; Thiemermann, C.; Brohi, K. Definition and drivers of acute traumatic coagulopathy: Clinical and experimental investigations. J. Thromb. Haemost. 2010, 8, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Yu, W.; Lin, Z.; Tan, S.; Bai, X.; Xu, L.; Dong, Y.; Li, N. A time course study of acute traumatic coagulopathy prior to resuscitation: From hypercoagulation to hypocoagulation caused by hypoperfusion? Transfus. Apher. Sci. 2014, 50, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Mohr, J.; Ruchholtz, S.; Hildebrand, F.; Flohé, S.; Frink, M.; Witte, I.; Weuster, M.; Fröhlich, M.; van Griensven, M.; Keibl, C.; et al. Induced hypothermia does not impair coagulation system in a swine multiple trauma model. J. Trauma Acute Care Surg. 2013, 74, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Tremoleda, J.L.; Watts, S.A.; Reynolds, P.S.; Thiemermann, C.; Brohi, K. Modeling Acute Traumatic Hemorrhagic Shock Injury: Challenges and Guidelines for Preclinical Studies. Shock 2017, 48, 610–623. [Google Scholar] [CrossRef]
- Gutierrez, G.; Reines, H.D.; Wulf-Gutierrez, M.E. Clinical review: Hemorrhagic shock. Crit. Care 2004, 8, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Fung, Y.L.; Tung, J.P.; Foley, S.R.; Simonova, G.; Thom, O.; Staib, A.; Collier, J.; Dunster, K.R.; Solano, C.; Shekar, K.; et al. Stored blood transfusion induces transient pulmonary arterial hypertension without impairing coagulation in an ovine model of nontraumatic haemorrhage. Vox Sang. 2013, 105, 150–158. [Google Scholar] [CrossRef]
- Krausz, M.M.; Bashenko, Y.; Hirsh, M. Crystalloid and colloid resuscitation of uncontrolled hemorrhagic shock following massive splenic injury. Shock 2001, 16, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Dyer, M.; Haldeman, S.; Gutierrez, A.; Kohut, L.; Sen Gupta, A.; Neal, M.D. Uncontrolled Hemorrhagic Shock Modeled via Liver Laceration in Mice with Real Time Hemodynamic Monitoring. J. Vis. Exp. 2017, e55554. [Google Scholar] [CrossRef] [Green Version]
- Sondeen, J.L.; Dubick, M.A.; Holcomb, J.B.; Wade, C.E. Uncontrolled hemorrhage differs from volume- or pressure-matched controlled hemorrhage in swine. Shock 2007, 28, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruscagin, V.; de Figueiredo, L.F.; Rasslan, S.; Varicoda, E.Y.; Rocha e Silva, M. Fluid resuscitation improves hemodynamics without increased bleeding in a model of uncontrolled hemorrhage induced by an iliac artery tear in dogs. J. Trauma 2002, 52, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Hirst, H.; Brinkman, J.; Beasley, A.; Crocker, R.; O’Sullivan, J. The effects of blood pressure on rebleeding when using ExcelArrest™ in a porcine model of lethal femoral injury. J. Emerg. Trauma Shock 2011, 4, 207–211. [Google Scholar] [CrossRef]
- Segura-Sampedro, J.J.; Pineño-Flores, C.; Craus-Miguel, A.; Morales-Soriano, R.; González-Argente, F.X. New hemostatic device for grade IV-V liver injury in porcine model: A proof of concept. World J. Emerg. Surg. 2019, 14, 58. [Google Scholar] [CrossRef]
- Darlington, D.N.; Gonzales, M.D.; Craig, T.; Dubick, M.A.; Cap, A.P.; Schwacha, M.G. Trauma-Induced Coagulopathy Is Associated with a Complex Inflammatory Response in the Rat. Shock 2015, 44 (Suppl. S1), 129–137. [Google Scholar] [CrossRef]
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.T.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.M.; Chen, S.H.; Rodor, J.; de Rooij, L.P.M.H.; Baker, A.H.; Carmeliet, P. Deciphering endothelial heterogeneity in health and disease at single-cell resolution: Progress and perspectives. Cardiovasc. Res. 2023, 119, 6–27. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Induction of Haemorrhage and/or Trauma | Species | Coagulopathic Changes | Endothelial Changes | Reference |
|---|---|---|---|---|
| Models of isolated haemorrhage | ||||
| Haemorrhage: fixed-volume (35% of total blood volume) | Pig | ↔ PT, ↔ aPTT, ↓ fibrinogen, ↑ fibrinogen degradation | - | Martini et al. [98] |
| Haemorrhage: fixed-volume (20% of total blood volume) | Rat | ↔ PT, ↔ aPTT, ↓ fibrinogen, ↑ factor II, ↔ factor V, ↑ factor X | - | Gangloff et al. [99] |
| Haemorrhage: pressure-controlled (35 mmHg for 90 min) | Mouse | - | ↓ syndecan-1 mRNA in lungs | Chipman et al. [100] |
| Haemorrhage: pressure-controlled (35 mmHg for 90 min) | Mouse | - | ↓ syndecan-1 mRNA in lungs | Wu et al. [101] |
| Haemorrhage: uncontrolled (tail amputation, liver punch biopsy, liver laceration, and spleen transection) | Rat | ↔ PT, ↔ aPTT, ↔ fibrinogen, ↔ FDPs, ↔ platelets | - | Morgan et al. [102] |
| Models of isolated trauma | ||||
| Trauma: Noble–Collip drum-induced blunt trauma | Rat | - | ↑ sTM, ↔ aPC, ↑ tPA, ↑ PAI-1, ↑ tPA-PAI-1 complex | Hayakawa et al. [103] |
| Trauma: Noble–Collip drum-induced blunt trauma | Rat | ↑ thrombin, ↑ soluble fibrin, ↓ platelets, ↓ antithrombin | - | Hayakawa et al. [104] |
| Trauma: closed mid-shaft limb fractures, laparotomy, and splenic crush | Rat | ↔ PT, ↔ aPTT, ↓ fibrinogen, ↑ factor II, ↔ factor V, ↑ factor X | - | Gangloff et al. [99] |
| Models of combined haemorrhage and trauma | ||||
| Haemorrhage: pressure-controlled (35 mmHg for 60 min) Trauma: laparotomy | Mouse | ↑ aPTT | ↑ aPC | Chesebro et al. [105] |
| Haemorrhage: pressure-controlled (35 mmHg for 90 min) Trauma: laparotomy | Mouse | - | ↓ VE-cadherin, ↓ ZO-1 | Barry et al. [106] |
| Haemorrhage: pressure-controlled (30 mmHg) Trauma: unilateral femur fracture with soft tissue damage | Pig | ↔ PT, ↔ aPTT, ↓ fibrinogen | - | White et al. [107] |
| Haemorrhage: pressure-controlled (35–40 mmHg for 60 min) Trauma: laparotomy | Rat | ↑ PT, ↑ aPTT, ↑ D-dimer, ↑ plasmin–antiplasmin complex | ↑ PAI-1, ↑ tPA, ↑ syndecan-1 | Xu et al. [108] |
| Models of combined haemorrhage and polytrauma | ||||
| Haemorrhage: pressure-controlled (30 mmHg for 90 min) Trauma: weight-drop-induced traumatic brain injury and laparotomy with bilateral abdominal rectus muscle crush injury | Mouse | - | ↑ syndecan-1, ↑ sTM | Wallen et al. [109] |
| Haemorrhage: pressure-controlled (40 mmHg) Trauma: right femur fracture and damage to small intestine, right and medial hepatic lobes, and skeletal muscle of right hind limb | Rat | ↑ PT, ↑ aPTT, ↓ fibrinogen, ↓ platelets, ↓ clot stability | - | Darlington et al. [110] |
| Haemorrhage: pressure-controlled haemorrhage (40 mmHg) Trauma: right femur fracture and damage to small intestines, left and medial hepatic lobes, and skeletal muscle of right hind limb | Rat | ↑ plasmin | ↑ TM, ↔ aPC, ↑ tPA, ↑ PAI-1 | Wu et al. [111] |
| Haemorrhage: fixed-volume (20% of total blood volume) Trauma: closed mid-shaft limb fractures, laparotomy, and splenic crush | Rat | ↑ PT, ↑ aPTT, ↓ fibrinogen, ↑ factor II, ↔ factor V, ↑ factor X | - | Gangloff et al. [99] |
| Haemorrhage: pressure-controlled (40–50 mmHg) Trauma: laparotomy and bilateral mid-shaft closed tibia and fibula fractures | Rat | ↓ fibrinogen, ↑ plasmi–antiplasmin complex | ↑ syndecan-1, ↑ aPC, ↑ sP-selectin | Xu et al. [112] |
| Haemorrhage: pressure-controlled (40–50 mmHg) Trauma: laparotomy and bilateral mid-shaft tibia and fibula fractures | Rat | ↑ PT, ↑ aPTT | - | Frith et al. [113] |
| Haemorrhage: pressure-controlled (40 mmHg) Trauma: small intestinal crush, liver injury, and right femur fracture | Pig | ↑ PT, ↓ fibrinogen, ↓ antithrombin III | - | Duan et al. [114] |
| Haemorrhage: pressure-controlled Trauma: blunt chest trauma and liver laceration | Pig | ↑ PT, ↓ fibrinogen | - | Mohr et al. [115] |
| Haemorrhage: pressure-controlled (30 mmHg) Trauma: right mid-shaft femur fracture and hind limb soft tissue injury | Pig | ↔ PT, ↔ aPTT, ↓ fibrinogen | - | White et al. [107] |
| Haemorrhage: pressure-controlled (25–30 mmHg for 60 min) Trauma: laparotomy and bilateral mid-shaft tibia and fibula fractures | Mouse | ↑ D-dimer, ↓ fibrinogen | ↑ aPC | Davenport et al. [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrett, L.; Curry, N.; Abu-Hanna, J. Experimental Models of Traumatic Injuries: Do They Capture the Coagulopathy and Underlying Endotheliopathy Induced by Human Trauma? Int. J. Mol. Sci. 2023, 24, 11174. https://doi.org/10.3390/ijms241311174
Barrett L, Curry N, Abu-Hanna J. Experimental Models of Traumatic Injuries: Do They Capture the Coagulopathy and Underlying Endotheliopathy Induced by Human Trauma? International Journal of Molecular Sciences. 2023; 24(13):11174. https://doi.org/10.3390/ijms241311174
Chicago/Turabian StyleBarrett, Liam, Nicola Curry, and Jeries Abu-Hanna. 2023. "Experimental Models of Traumatic Injuries: Do They Capture the Coagulopathy and Underlying Endotheliopathy Induced by Human Trauma?" International Journal of Molecular Sciences 24, no. 13: 11174. https://doi.org/10.3390/ijms241311174