
Deep Characterization of a Greek Patient with Desmin-Related Myofibrillar Myopathy and Cardiomyopathy


 , , , ,
, , , ,
Abstract
:1. Introduction
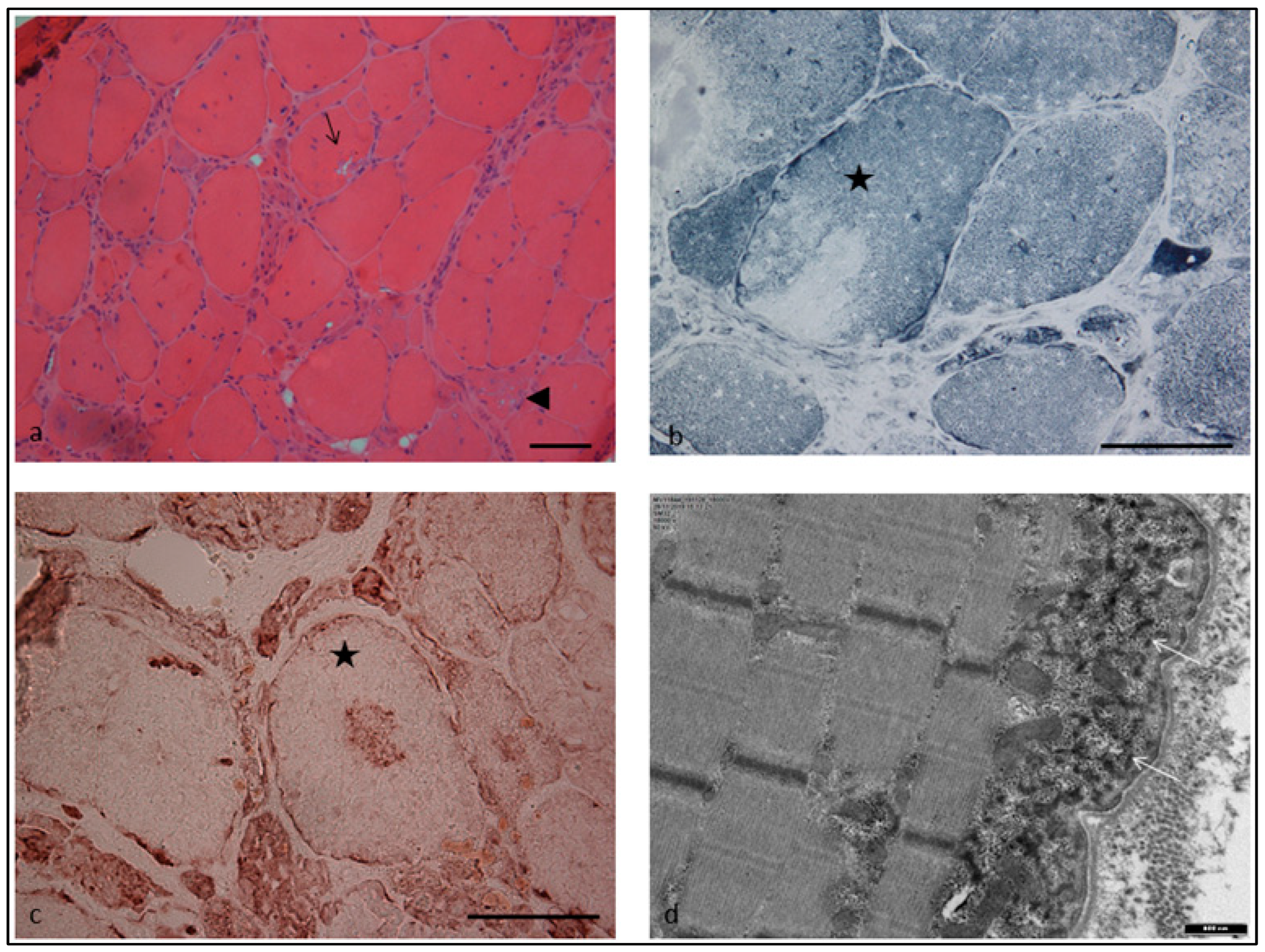
2. Case Report
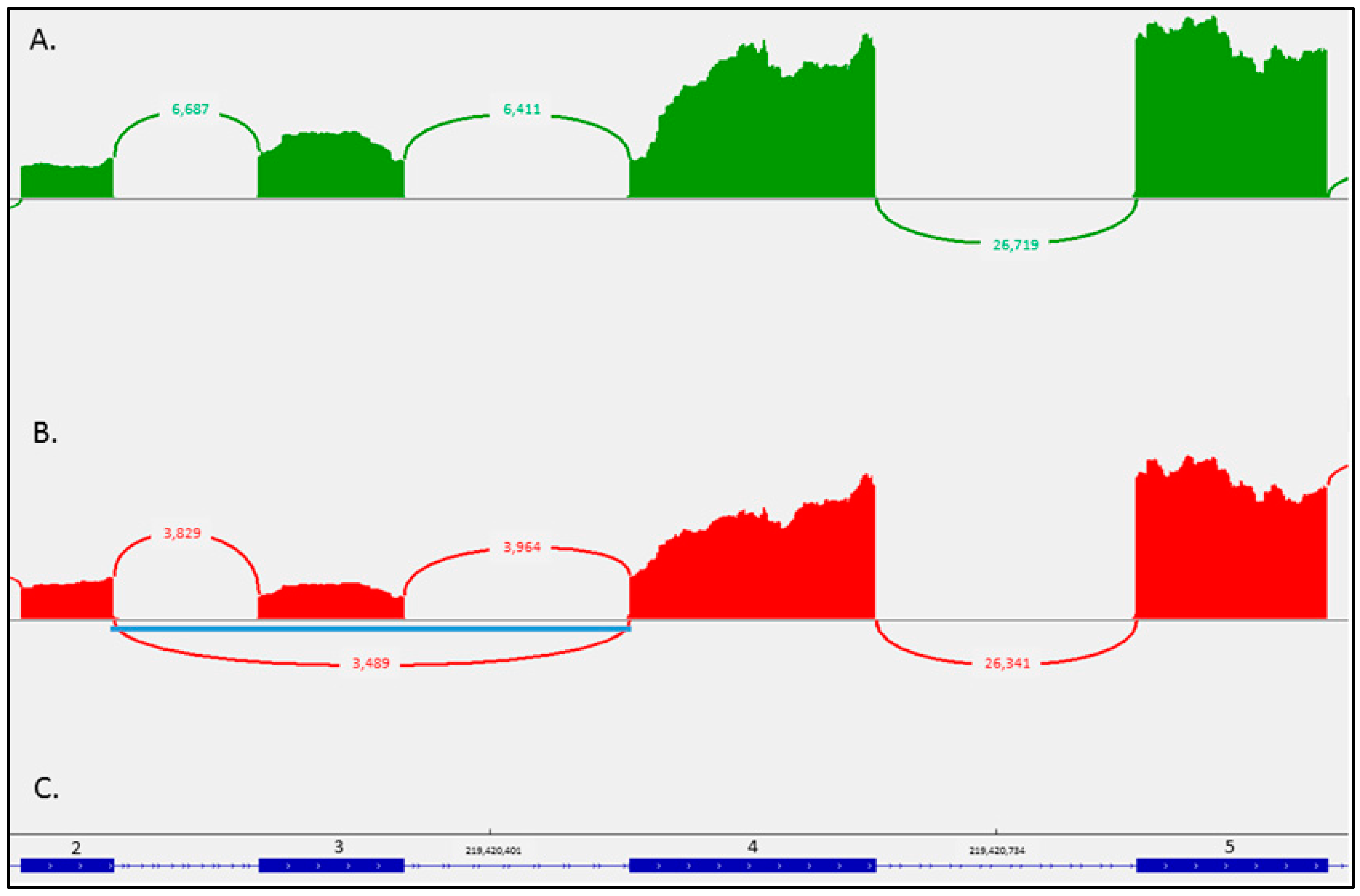
3. Discussion
4. Materials and Methods
4.1. Muscle Biopsy
4.2. Targeted Gene Enrichment, Next Generation Sequencing NGS (High-Throughput Sequencing)
4.3. Bioinformatics Analysis
4.4. Variants Interpretations
4.5. RNA Sequencing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clemen, C.S.; Herrmann, H.; Strelkov, S.V.; Schröder, R. Desminopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 47–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, E.; Weber, K. Intermediate filaments: Structure, dynamics, function and disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; Fardeau, M. Myofibrillar myopathies. Handb. Clin. Neurol. 2013, 113, 1337–1342. [Google Scholar] [CrossRef]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [Green Version]
- Olivé, M.; Kley, R.A.; Goldfarb, L.G. Myofibrillar myopathies: New developments. Curr. Opin. Neurol. 2013, 26, 527–535. [Google Scholar] [CrossRef]
- Olivé, M.; Odgerel, Z.; Martínez, A.; Poza, J.J.; Bragado, F.G.; Zabalza, R.J.; Jericó, I.; Gonzalez-Mera, L.; Shatunov, A.; Lee, H.S.; et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul. Disord. 2011, 21, 533–542. [Google Scholar] [CrossRef] [Green Version]
- van Spaendonck-Zwarts, K.; van Hessem, L.; Jongbloed, J.; de Walle, H.; Capetanaki, Y.; van der Kooi, A.; van Langen, I.; Berg, M.v.D.; van Tintelen, J. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef]
- Walter, M.C.; Reilich, P.; Huebner, A.; Fischer, D.; Schroder, R.; Vorgerd, M.; Kress, W.; Born, C.; Schoser, B.; Krause, K.H.; et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain 2007, 130, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated with a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef]
- Oka, H.; Nakau, K.; Imanishi, R.; Furukawa, T.; Tanabe, Y.; Hirono, K.; Hata, Y.; Nishida, N.; Azuma, H. A Case Report of a Rare Heterozygous Variant in the Desmin Gene Associated with Hypertrophic Cardiomyopathy and Complete Atrioventricular Block. CJC Open 2021, 3, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tapscoft, T.; Gonzalez, O.; Burch, P.E.; Quiñones, M.A.; Zoghbi, W.A.; Hill, R.; Bachinski, L.L.; Mann, D.; Roberts, R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999, 100, 461–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruszczyk, P.; Kostera-Pruszczyk, A.; Shatunov, A.; Goudeau, B.; Dramiñska, A.; Takeda, K.; Sambuughin, N.; Vicart, P.; Strelkov, S.V.; Goldfarb, L.G.; et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int. J. Cardiol. 2007, 117, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gärtner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Schröder, R.; Schoser, B. Myofibrillar myopathies: A clinical and myopathological guide. Brain Pathol. 2009, 19, 483–492. [Google Scholar] [CrossRef]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Wahbi, K.; Béhin, A.; Charron, P.; Dunand, M.; Richard, P.; Meune, C.; Vicart, P.; Laforêt, P.; Stojkovic, T.; Bécane, H.M.; et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul. Disord. 2012, 22, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Béhin, A.; Salort-Campana, E.; Wahbi, K.; Richard, P.; Carlier, R.-Y.; Carlier, P.; Laforêt, P.; Stojkovic, T.; Maisonobe, T.; Verschueren, A.; et al. Myofibrillar myopathies: State of the art, present and future challenges. Rev. Neurol. 2015, 171, 715–729. [Google Scholar] [CrossRef]
- Dagvadorj, A.; Goudeau, B.; Hilton-Jones, D.; Blancato, J.K.; Shatunov, A.; Simon-Casteras, M.; Squier, W.; Nagle, J.W.; Goldfarb, L.G.; Vicart, P. Respiratory insufficiency in desminopathy patients caused by introduction of proline residues in desmin c-terminal alpha-helical segment. Muscle Nerve 2003, 27, 669–675. [Google Scholar] [CrossRef]
- Schramm, N.; Born, C.; Weckbach, S.; Reilich, P.; Walter, M.C.; Reiser, M.F. Involvement patterns in myotilinopathy and desminopathy detected by a novel neuromuscular whole-body MRI protocol. Eur. Radiol. 2008, 18, 2922–2936. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; van der Ven, P.F.M.; Behin, A.; Stojkovic, T.; Eymard, B.; Dubourg, O.; Laforêt, P.; Faulkner, G.; Richard, P.; Vicart, P.; et al. Differential involvement of sarcomeric proteins in myofibrillar myopathies: A morphological and immunohistochemical study. Acta Neuropathol. 2009, 117, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Park, K.-Y.; Semino-Mora, C.; Lee, H.S.; Sivakumar, K.; Goldfarb, L.G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. New Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef]
- Ojrzyńska, N.; Bilińska, Z.T.; Franaszczyk, M.; Płoski, R.; Grzybowski, J. Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol. Polska 2017, 75, 723. [Google Scholar] [CrossRef] [Green Version]
- Kostareva, A.; Gudkova, A.; Sjoberg, G.; Kiselev, I.; Moiseeva, O.; Karelkina, E.; Goldfarb, L.; Schlyakhto, E.; Sejersen, T. Desmin mutations in a St. Petersburg cohort of cardiomyopathies. Acta Myol. 2006, 25, 109–115. [Google Scholar]
- Fan, P.; Lu, C.-X.; Dong, X.-Q.; Zhu, D.; Yang, K.-Q.; Liu, K.-Q.; Zhang, D.; Zhang, Y.; Meng, X.; Tan, H.-Q.; et al. A novel phenotype with splicing mutation identified in a Chinese family with desminopathy. Chin. Med. J. 2019, 132, 127–134. [Google Scholar] [CrossRef]
- Nalini, A.; Richard, P.; Urtizberea, J.; Cobo, A.-M.; Gayathri, N. New mutation of the desmin gene identified in an extended Indian pedigree presenting with distal myopathy and cardiac disease. Neurol. India 2013, 61, 622. [Google Scholar] [CrossRef]
- Arbustini, E.; Di Toro, A.; Giuliani, L.; Favalli, V.; Narula, N.; Grasso, M. Cardiac Phenotypes in Hereditary Muscle Disorders. J. Am. Coll. Cardiol. 2018, 72, 2485–2506. [Google Scholar] [CrossRef]
- Kostera-Pruszczyk, A.; Pruszczyk, P.; Kamińska, A.; Lee, H.-S.; Goldfarb, L.G. Diversity of cardiomyopathy phenotypes caused by mutations in desmin. Int. J. Cardiol. 2008, 131, 146–147. [Google Scholar] [CrossRef]
- Strach, K.; Sommer, T.; Grohé, C.; Meyer, C.; Fischer, D.; Walter, M.C.; Vorgerd, M.; Reilich, P.; Bär, H.; Reimann, J.; et al. Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul. Disord. 2008, 18, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.G. Magnetic resonance imaging patterns of muscle involvement in genetic muscle diseases: A systematic review. J. Neurol. 2016, 264, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Kley, R.A.; Strach, K.; Meyer, C.; Sommer, T.; Eger, K.; Rolfs, A.; Meyer, W.; Pou, A.; Pradas, J.; et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 2008, 71, 758–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claeys, K.; Fardeau, M.; Schröder, R.; Suominen, T.; Tolksdorf, K.; Béhin, A.; Dubourg, O.; Eymard, B.; Maisonobe, T.; Stojkovic, T.; et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul. Disord. 2008, 18, 656–666. [Google Scholar] [CrossRef]
- Vrabie, A.; Goldfarb, L.G.; Shatunov, A.; Nägele, A.; Fritz, P.; Kaczmarek, I.; Goebel, H.H. The enlarging spectrum of desminopathies: New morphological findings, eastward geographic spread, novel exon 3 desmin mutation. Acta Neuropathol. 2005, 109, 411–417. [Google Scholar] [CrossRef]
- Park, K.Y.; Dalakas, M.C.; Goebel, H.H.; Ferrans, V.J.; Semino-Mora, C.; Litvak, S.; Takeda, K.; Goldfarb, L.G. Desmin splice variants causing cardiac and skeletal myopathy. J. Med. Genet. 2000, 37, 851–857. [Google Scholar] [CrossRef]
- Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Körperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3. Biomedicines 2021, 9, 1400. [Google Scholar] [CrossRef]
- Bang, M.-L.; Gregorio, C.; Labeit, S. Molecular Dissection of the Interaction of Desmin with the C-Terminal Region of Nebulin. J. Struct. Biol. 2002, 137, 119–127. [Google Scholar] [CrossRef]
- Krahn, M.; Biancalana, V.; Cerino, M.; Perrin, A.; Michel-Calemard, L.; Nectoux, J.; Leturcq, F.; Bouchet-Séraphin, C.; Acquaviva-Bourdain, C.; Campana-Salort, E.; et al. A National French consensus on gene lists for the diagnosis of myopathies using next-generation sequencing. Eur. J. Hum. Genet. 2019, 27, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| ACTA1 (NM_001100.3) |
| BAG3 (NM_004281.3) |
| CRYAB (NM_001885.2) |
| DES (NM_001927.3) |
| DNAJB6 (NM_058246.3) |
| FHL1 (NM_001159702.2) |
| FLNC (NM_001458.4) |
| GNE (NM_001128227.3) |
| HSPB1 (NM_001540.3) |
| HSPB8 (NM_014365.2) |
| MYH2 (NM_017534.5) |
| MYOT (NM_006790.2) |
| RYR1 (NM_000540.2, with partially covered exon 91) |
| SQSTM1 (NM_003900.4) |
| TTN (NM_001267550.1) |
| VCP (NM_007126.3) |
| ZASP/LDB3 (NM_001080114.1, NM_007078.2, NM_001171610.1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulos, C.; Malfatti, E.; Métay, C.; Keren, B.; Lejeune, E.; Buratti, J.; Xirou, S.; Chrysanthou-Piterou, M.; Papadimas, G.K. Deep Characterization of a Greek Patient with Desmin-Related Myofibrillar Myopathy and Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 11181. https://doi.org/10.3390/ijms241311181
Papadopoulos C, Malfatti E, Métay C, Keren B, Lejeune E, Buratti J, Xirou S, Chrysanthou-Piterou M, Papadimas GK. Deep Characterization of a Greek Patient with Desmin-Related Myofibrillar Myopathy and Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(13):11181. https://doi.org/10.3390/ijms241311181
Chicago/Turabian StylePapadopoulos, Constantinos, Edoardo Malfatti, Corinne Métay, Boris Keren, Elodie Lejeune, Julien Buratti, Sophia Xirou, Margarita Chrysanthou-Piterou, and George K. Papadimas. 2023. "Deep Characterization of a Greek Patient with Desmin-Related Myofibrillar Myopathy and Cardiomyopathy" International Journal of Molecular Sciences 24, no. 13: 11181. https://doi.org/10.3390/ijms241311181
APA StylePapadopoulos, C., Malfatti, E., Métay, C., Keren, B., Lejeune, E., Buratti, J., Xirou, S., Chrysanthou-Piterou, M., & Papadimas, G. K. (2023). Deep Characterization of a Greek Patient with Desmin-Related Myofibrillar Myopathy and Cardiomyopathy. International Journal of Molecular Sciences, 24(13), 11181. https://doi.org/10.3390/ijms241311181