
The Role of Autophagy and Apoptosis in Affected Skin and Lungs in Patients with Systemic Sclerosis
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Fibroblasts—Function and Transformation into Myofibroblasts in SSc
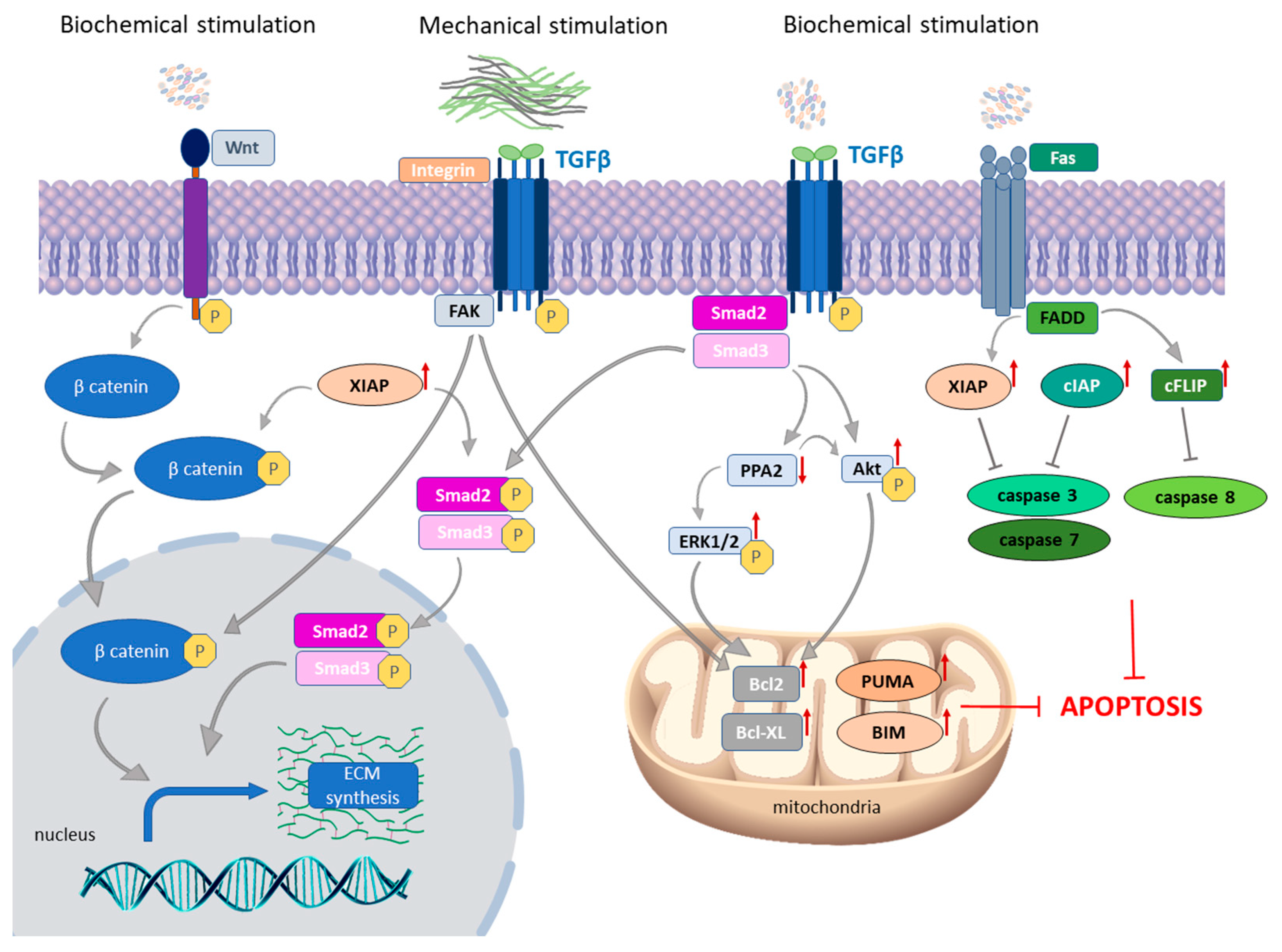
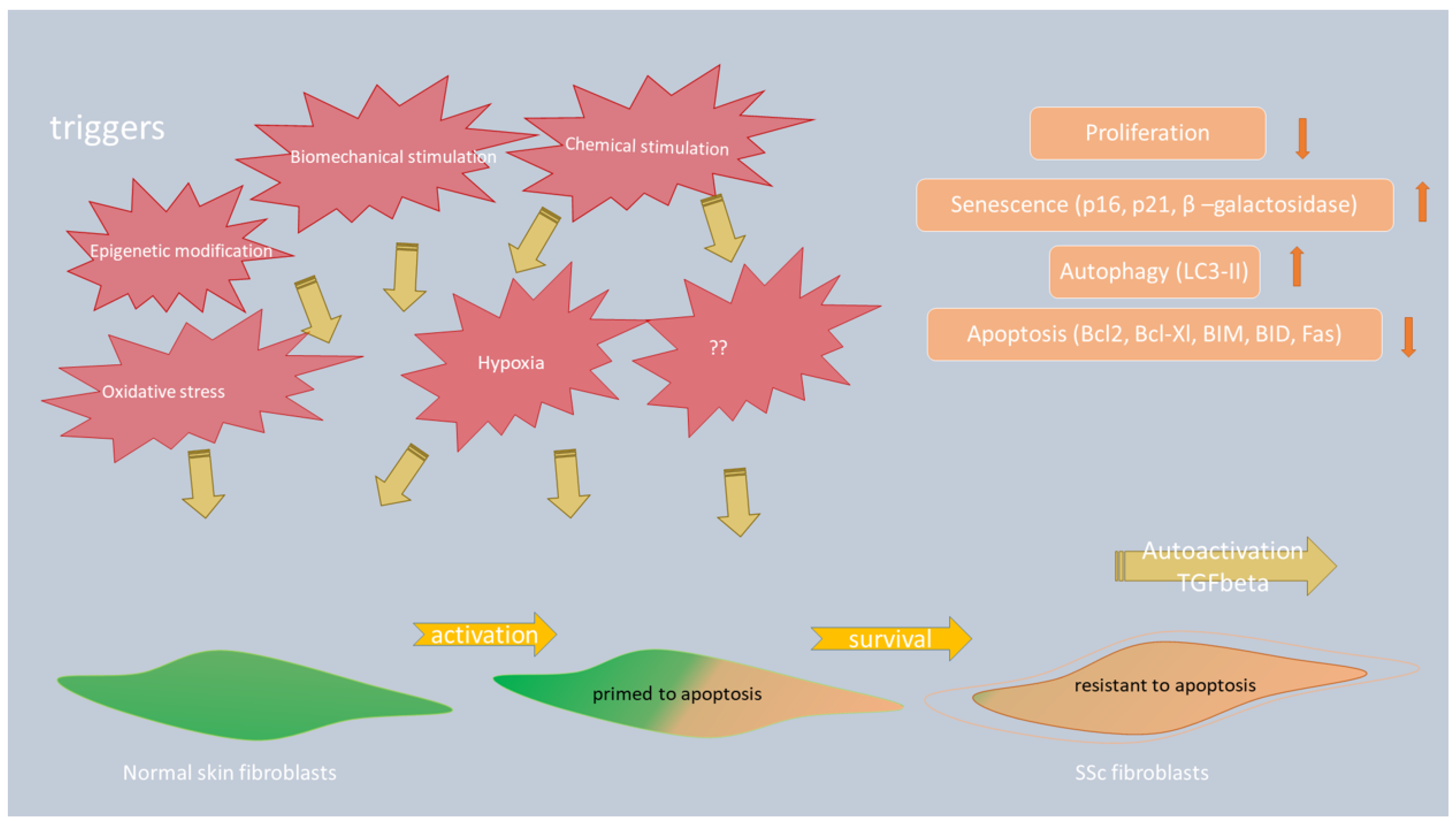
3.1. The Role of Apoptosis in Myofibroblast Survival in SSc
3.1.1. TGF-β as a Crucial Molecule in Fibroblast-to-Myofibroblast Transition in SSc Skin
3.1.2. Autocrine TGF-β Signaling and Death Receptor Apoptotic Pathway
3.1.3. The Link between Intrinsic and Extrinsic Apoptotic Pathways in SSc Skin
3.1.4. The Role of Mechanical Stimulation on the Expression of Apoptotic Proteins in SSc Fibroblasts
3.2. The Role of Autophagy in the Progression of Skin Fibrosis in Patients with SSc
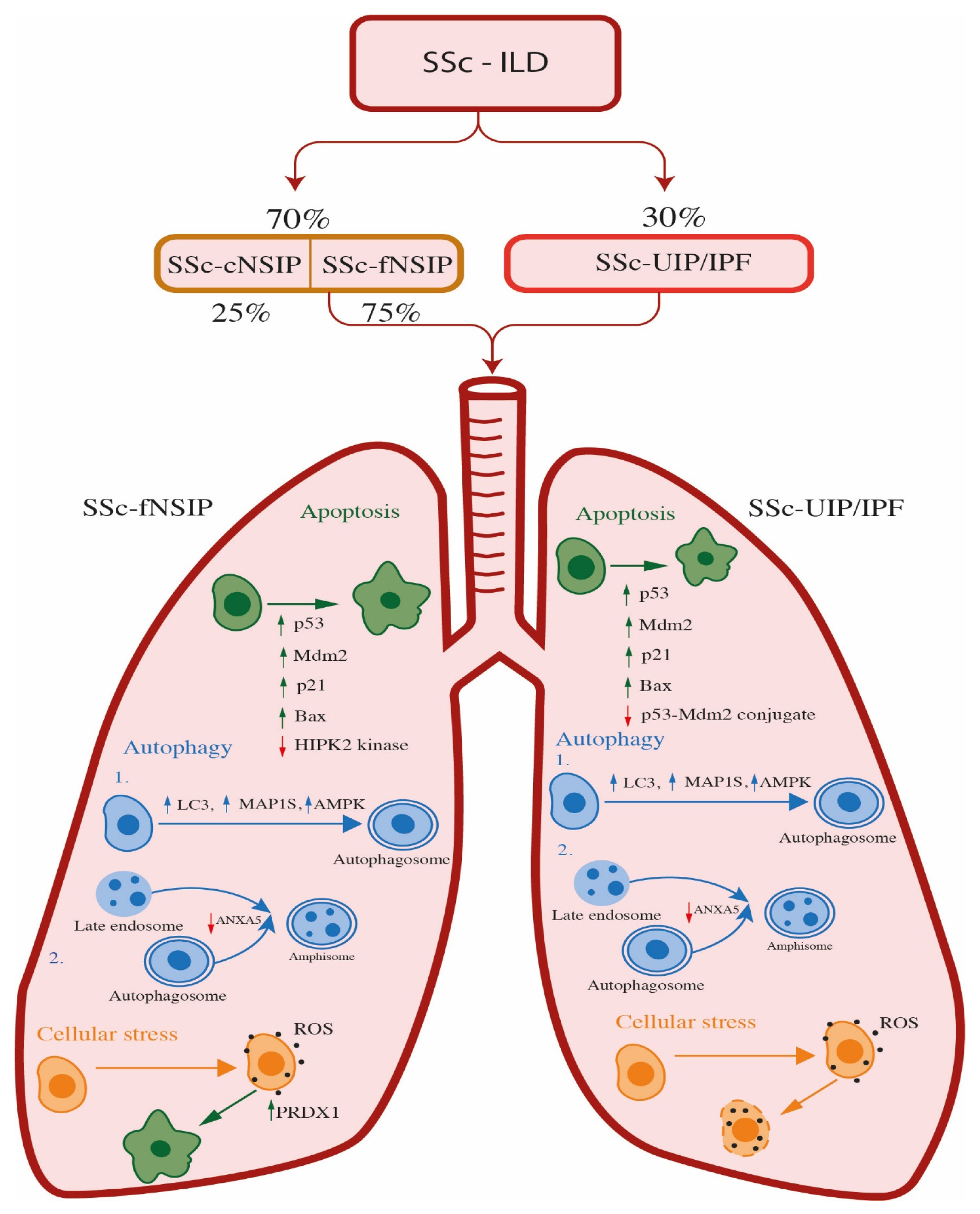
4. Systemic Sclerosis—Interstitial Lung Disease (SSc-ILD)
4.1. The Role of Apoptosis and Oxidative Stress in SSc-ILD Patients
4.2. The Role of Autophagy in SSc-ILD Patients
4.2.1. The Role of Autophagy in SSc-ILD Patients with UIP Pattern
4.2.2. The Role of Autophagy in SSc-ILD Patients with fNSIP Pattern
5. Conclusions
6. Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muangchan, C.; Canadian Scleroderma Research Group; Baron, M.; Pope, J. The 15% Rule in Scleroderma: The Frequency of Severe Organ Complications in Systemic Sclerosis. A Systematic Review. J. Rheumatol. 2013, 40, 1545–1556. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S.; Rose, N.R. Sex Differences in Autoimmune Disease from a Pathological Perspective. Am. J. Pathol. 2008, 173, 600–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Ramelius, A.; Barton, J.; et al. Activating Germline Mutations in STAT3 Cause Early-Onset Multi-Organ Autoimmune Disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef]
- Amur, S.; Parekh, A.; Mummaneni, P. Sex Differences and Genomics in Autoimmune Diseases. J. Autoimmun. 2012, 38, J254–J265. [Google Scholar] [CrossRef]
- Czirjak, L.; Foeldvari, I.; Muller-Ladner, U. Skin Involvement in Systemic Sclerosis. Rheumatology 2008, 47, v44–v45. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, M.; Abuchar, A.; Santana, N.; Dehesa, L.; Kerdel, F.A. An Update on the Treatment of the Cutaneous Manifestations of Systemic Sclerosis: The Dermatologist’s Point of View. J. Clin. Aesthetic Dermatol. 2012, 5, 33–43. [Google Scholar]
- Van Den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A.; Carreira, P.E.; et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League against Rheumatism Collaborative Initiative. Ann. Rheum. Dis. 2013, 72, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR Recommendations for the Treatment of Systemic Sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef]
- Suliman, S.; Al Harash, A.; Roberts, W.N.; Perez, R.L.; Roman, J. Scleroderma-Related Interstitial Lung Disease. Respir. Med. Case Rep. 2017, 22, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Lancet, T. Systemic Sclerosis: Advances and Prospects. Lancet 2017, 390, 1624. [Google Scholar] [CrossRef] [Green Version]
- De Almeida Chaves, S.; Porel, T.; Mounié, M.; Alric, L.; Astudillo, L.; Huart, A.; Lairez, O.; Michaud, M.; Prévot, G.; Ribes, D.; et al. Sine Scleroderma, Limited Cutaneous, and Diffused Cutaneous Systemic Sclerosis Survival and Predictors of Mortality. Arthritis Res. Ther. 2021, 23, 295. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, W.A.; Fries, J.F.; Masi, A.T.; Shulman, L.E. Pathologic Observations in Systemic Sclerosis (Scleroderma). A Study of Fifty-Eight Autopsy Cases and Fifty-Eight Matched Controls. Am. J. Med. 1969, 46, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, Diagnosis and Clinical Course of the Spectrum of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef]
- Cutolo, M.; Soldano, S.; Smith, V. Pathophysiology of Systemic Sclerosis: Current Understanding and New Insights. Expert Rev. Clin. Immunol. 2019, 15, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Wei, J.; Varga, J. Understanding Fibrosis in Systemic Sclerosis: Shifting Paradigms, Emerging Opportunities. Nat. Rev. Rheumatol. 2011, 8, 42–54. [Google Scholar] [CrossRef]
- Varga, J.A.; Trojanowska, M. Fibrosis in Systemic Sclerosis. Rheum. Dis. Clin. N. Am. 2008, 34, 115–143. [Google Scholar] [CrossRef]
- Wu, D.J.; Adamopoulos, I.E. Autophagy and Autoimmunity. Clin. Immunol. 2017, 176, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Thorburn, A. Apoptosis and Autophagy: Regulatory Connections between Two Supposedly Different Processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Del Principe, D.; Vona, R.; Giordani, L.; Straface, E.; Giammarioli, A.M. Defective Autophagy in Fibroblasts May Contribute to Fibrogenesis in Autoimmune Processes. Curr. Pharm. Des. 2011, 17, 3878–3887. [Google Scholar] [CrossRef]
- Principe, D.D.; Lista, P.; Malorni, W.; Giammarioli, A.M. Fibroblast Autophagy in Fibrotic Disorders. J. Pathol. 2013, 229, 208–220. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-Dependent Cell Death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Zochling, J.; Newell, F.; Charlesworth, J.C.; Leo, P.; Stankovich, J.; Cortes, A.; Zhou, Y.; Stevens, W.; Sahhar, J.; Roddy, J.; et al. An Immunochip-Based Interrogation of Scleroderma Susceptibility Variants Identifies a Novel Association at DNASE1L3. Arthritis Res. 2014, 16, 438. [Google Scholar] [CrossRef] [Green Version]
- Mayes, M.D.; Bossini-Castillo, L.; Gorlova, O.; Martin, J.E.; Zhou, X.; Chen, W.V.; Assassi, S.; Ying, J.; Tan, F.K.; Arnett, F.C.; et al. Immunochip Analysis Identifies Multiple Susceptibility Loci for Systemic Sclerosis. Am. J. Hum. Genet. 2014, 94, 47–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoy, E.C.; Black, C.; Fleischmajer, R.; Jablonska, S.; Krieg, T.; Medsger, T.A.; Rowell, N.; Wollheim, F. Scleroderma (Systemic Sclerosis): Classification, Subsets and Pathogenesis. J. Rheumatol. 1988, 15, 202–205. [Google Scholar]
- Di Battista, M.; Lepri, G.; Codullo, V.; Da Rio, M.; Fiorentini, E.; Della Rossa, A.; Guiducci, S. Systemic Sclerosis: One Year in Review 2023. Clin. Exp. Rheumatol. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and Mechano-Regulation of Connective Tissue Remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of Endothelial-Mesenchymal Transition (EndoMT) in the Pathogenesis of Fibrotic Disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Hao, H.; Gabbiani, G.; Camenzind, E.; Bacchetta, M.; Virmani, R.; Bochaton-Piallat, M.-L. Phenotypic Modulation of Intima and Media Smooth Muscle Cells in Fatal Cases of Coronary Artery Lesion. Arter. Thromb. Vasc. Biol. 2006, 26, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, V.S.; Howell, K.; Csiszar, K.; Denton, C.P.; Black, C.M.; Abraham, D.J. Shared Expression of Phenotypic Markers in Systemic Sclerosis Indicates a Convergence of Pericytes and Fibroblasts to a Myofibroblast Lineage in Fibrosis. Arthritis Res. Ther. 2005, 7, R1113–R1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in Murine Cutaneous Fibrosis Originate from Adiponectin-Positive Intradermal Progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, A.-H.; Schönborn, K.; Krieg, T. Pathophysiology of Systemic Sclerosis (Scleroderma). Kaohsiung J. Med. Sci. 2022, 38, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Castello-Cros, R.; Whitaker-Menezes, D.; Molchansky, A.; Purkins, G.; Soslowsky, L.J.; Beason, D.P.; Sotgia, F.; Iozzo, R.V.; Lisanti, M.P. Scleroderma-like Properties of Skin from Caveolin-1-Deficient Mice: Implications for New Treatment Strategies in Patients with Fibrosis and Systemic Sclerosis. Cell Cycle 2011, 10, 2140–2150. [Google Scholar] [CrossRef] [Green Version]
- Chabaud, S.; Moulin, V.J. Apoptosis Modulation as a Promising Target for Treatment of Systemic Sclerosis. Int. J. Rheumatol. 2011, 2011, 495792. [Google Scholar] [CrossRef] [Green Version]
- Jelaska, A.; Korn, J.H. Role of Apoptosis and Transforming Growth Factor Beta1 in Fibroblast Selection and Activation in Systemic Sclerosis. Arthritis Rheum. 2000, 43, 2230–2239. [Google Scholar] [CrossRef]
- Kissin, E.; Korn, J.H. Apoptosis and Myofibroblasts in the Pathogenesis of Systemic Sclerosis. Curr. Rheumatol. Rep. 2002, 4, 129–135. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nishioka, K. Possible Role of Apoptosis in the Pathogenesis of Bleomycin-Induced Scleroderma. J. Investig. Dermatol. 2004, 122, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagares, D.; Santos, A.; Grasberger, P.E.; Liu, F.; Probst, C.K.; Rahimi, R.A.; Sakai, N.; Kuehl, T.; Ryan, J.; Bhola, P.; et al. Targeted Apoptosis of Myofibroblasts with the BH3 Mimetic ABT-263 Reverses Established Fibrosis. Sci. Transl. Med. 2017, 9, eaal3765. [Google Scholar] [CrossRef]
- Chabaud, S.; Corriveau, M.-P.; Grodzicky, T.; Senécal, J.-L.; Chartier, S.; Raymond, Y.; Moulin, V.J. Decreased Secretion of MMP by Non-Lesional Late-Stage Scleroderma Fibroblasts after Selection via Activation of the Apoptotic Fas-Pathway. J. Cell. Physiol. 2011, 226, 1907–1914. [Google Scholar] [CrossRef]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative Stress in Cancer and Fibrosis: Opportunity for Therapeutic Intervention with Antioxidant Compounds, Enzymes, and Nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Zhang, W.; Zhang, L. MicroRNAs Play an Essential Role in Autophagy Regulation in Various Disease Phenotypes. BioFactors 2019, 45, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, Y.; Wen, D.; Wang, J. Noncoding RNAs: Master Regulator of Fibroblast to Myofibroblast Transition in Fibrosis. Int. J. Mol. Sci. 2023, 24, 1801. [Google Scholar] [CrossRef] [PubMed]
- Thoreau, B.; Chaigne, B.; Renaud, A.; Mouthon, L. Pathophysiology of Systemic Sclerosis. La Presse Médicale 2021, 50, 104087. [Google Scholar] [CrossRef]
- Ihn, H. The Role of TGF-Beta Signaling in the Pathogenesis of Fibrosis in Scleroderma. Arch. Immunol. Ther. Exp. 2002, 50, 325–331. [Google Scholar]
- Ihn, H. Pathogenesis of Fibrosis: Role of TGF-Beta and CTGF. Curr. Opin. Rheumatol. 2002, 14, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X.-J. Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
- Bergmann, C.; Distler, J.H.W. Canonical Wnt Signaling in Systemic Sclerosis. Lab. Investig. 2016, 96, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Hallenberger, L.; Chenguiti Fakhouri, S.; Merlevede, B.; Brandt, A.; Dees, C.; Zhu, H.; Zehender, A.; Zhou, X.; Schwab, A.; et al. X-Linked Inhibitor of Apoptosis Protein (XIAP) Inhibition in Systemic Sclerosis (SSc). Ann. Rheum. Dis. 2021, 80, 1048–1056. [Google Scholar] [CrossRef]
- Kawakami, T.; Ihn, H.; Xu, W.; Smith, E.; LeRoy, C.; Trojanowska, M. Increased Expression of TGF-Beta Receptors by Scleroderma Fibroblasts: Evidence for Contribution of Autocrine TGF-Beta Signaling to Scleroderma Phenotype. J. Investig. Dermatol. 1998, 110, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Kubo, M.; Ihn, H.; Yamane, K.; Tamaki, K. Upregulated Expression of Transforming Growth Factor-Beta Receptors in Dermal Fibroblasts of Skin Sections from Patients with Systemic Sclerosis. J. Rheumatol. 2002, 29, 2558–2564. [Google Scholar] [PubMed]
- Yamane, K.; Ihn, H.; Kubo, M.; Tamaki, K. Increased Transcriptional Activities of Transforming Growth Factor Beta Receptors in Scleroderma Fibroblasts. Arthritis Rheum. 2002, 46, 2421–2428. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.A.; Thatcher, T.H.; Olsen, K.C.; Maggirwar, S.B.; Phipps, R.P.; Sime, P.J. PPAR-γ Ligands Repress TGFβ-Induced Myofibroblast Differentiation by Targeting the PI3K/Akt Pathway: Implications for Therapy of Fibrosis. PLoS ONE 2011, 6, e15909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Chen, S.-J.; Wu, M.; Warner-Blankenship, M.; Ning, H.; Lakos, G.; Mori, Y.; Chang, E.; Nihijima, C.; Takehara, K.; et al. Smad-Independent Transforming Growth Factor-Beta Regulation of Early Growth Response-1 and Sustained Expression in Fibrosis: Implications for Scleroderma. Am. J. Pathol. 2008, 173, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Pannu, J.; Nakerakanti, S.; Smith, E.; ten Dijke, P.; Trojanowska, M. Transforming Growth Factor-Beta Receptor Type I-Dependent Fibrogenic Gene Program Is Mediated via Activation of Smad1 and ERK1/2 Pathways. J. Biol. Chem. 2007, 282, 10405–10413. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Cawley, N.X.; Loh, Y.P. Carboxypeptidase E/NFα1: A New Neurotrophic Factor against Oxidative Stress-Induced Apoptotic Cell Death Mediated by ERK and PI3-K/AKT Pathways. PLoS ONE 2013, 8, e71578. [Google Scholar] [CrossRef] [Green Version]
- Samuel, G.H.; Bujor, A.M.; Nakerakanti, S.S.; Hant, F.N.; Trojanowska, M. Autocrine Transforming Growth Factor β Signaling Regulates Extracellular Signal-Regulated Kinase 1/2 Phosphorylation via Modulation of Protein Phosphatase 2A Expression in Scleroderma Fibroblasts. Fibrogenesis Tissue Repair 2010, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Augé, N.; Andrieu, N.; Nègre-Salvayre, A.; Thiers, J.-C.; Levade, T.; Salvayre, R. The Sphingomyelin-Ceramide Signaling Pathway Is Involved in Oxidized Low Density Lipoprotein-Induced Cell Proliferation. J. Biol. Chem. 1996, 271, 19251–19255. [Google Scholar] [CrossRef] [Green Version]
- Samuel, G.H.; Lenna, S.; Bujor, A.M.; Lafyatis, R.; Trojanowska, M. Acid Sphingomyelinase Deficiency Contributes to Resistance of Scleroderma Fibroblasts to Fas-Mediated Apoptosis. J. Dermatol. Sci. 2012, 67, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Meier, P. Inhibitor of Apoptosis (IAP) Proteins-Modulators of Cell Death and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef] [Green Version]
- Dumétier, B.; Zadoroznyj, A.; Dubrez, L. IAP-Mediated Protein Ubiquitination in Regulating Cell Signaling. Cells 2020, 9, 1118. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Denault, J.-B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. XIAP Inhibits Caspase-3 and -7 Using Two Binding Sites: Evolutionarily Conserved Mechanism of IAPs. EMBO J. 2005, 24, 645–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, C.; Schramm, A.; Akhmetshina, A.; Dees, C.; Kireva, T.; Gelse, K.; Sonnylal, S.; De Crombrugghe, B.; Taketo, M.M.; Distler, O.; et al. β-Catenin Is a Central Mediator of pro-Fibrotic Wnt Signaling in Systemic Sclerosis. Ann. Rheum. Dis. 2012, 71, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/β-Catenin Signaling Is Hyperactivated in Systemic Sclerosis and Induces Smad-Dependent Fibrotic Responses in Mesenchymal Cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback Amplification of Fibrosis through Matrix Stiffening and COX-2 Suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic Extracellular Matrix Activates a Profibrotic Positive Feedback Loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [Green Version]
- Shi-wen, X.; Thompson, K.; Khan, K.; Liu, S.; Murphy-Marshman, H.; Baron, M.; Denton, C.P.; Leask, A.; Abraham, D.J. Focal Adhesion Kinase and Reactive Oxygen Species Contribute to the Persistent Fibrotic Phenotype of Lesional Scleroderma Fibroblasts. Rheumatology 2012, 51, 2146–2154. [Google Scholar] [CrossRef] [Green Version]
- Klapan, K.; Simon, D.; Karaulov, A.; Gomzikova, M.; Rizvanov, A.; Yousefi, S.; Simon, H.-U. Autophagy and Skin Diseases. Front. Pharmacol. 2022, 13, 844756. [Google Scholar] [CrossRef]
- Nagar, R. Autophagy: A Brief Overview in Perspective of Dermatology. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 290. [Google Scholar] [CrossRef]
- Frech, T.; De Domenico, I.; Murtaugh, M.A.; Revelo, M.P.; Li, D.Y.; Sawitzke, A.D.; Drakos, S. Autophagy Is a Key Feature in the Pathogenesis of Systemic Sclerosis. Rheumatol. Int. 2014, 34, 435–439. [Google Scholar] [CrossRef]
- Dumit, V.I.; Küttner, V.; Käppler, J.; Piera-Velazquez, S.; Jimenez, S.A.; Bruckner-Tuderman, L.; Uitto, J.; Dengjel, J. Altered MCM Protein Levels and Autophagic Flux in Aged and Systemic Sclerosis Dermal Fibroblasts. J. Investig. Dermatol. 2014, 134, 2321–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Tamura, N.; Waguri, S.; Yamamoto, T. Autophagy Is Involved in the Sclerotic Phase of Systemic Sclerosis. FJMS 2020, 66, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perelas, A.; Silver, R.M.; Arrossi, A.V.; Highland, K.B. Systemic Sclerosis-Associated Interstitial Lung Disease. Lancet Respir. Med. 2020, 8, 304–320. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Changes in Causes of Death in Systemic Sclerosis, 1972–2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef]
- Gilson, M.; Zerkak, D.; Wipff, J.; Dusser, D.; Dinh-Xuan, A.T.; Abitbol, V.; Chaussade, S.; Legmann, P.; Kahan, A.; Allanore, Y. Prognostic Factors for Lung Function in Systemic Sclerosis: Prospective Study of 105 Cases. Eur. Respir. J. 2010, 35, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Nihtyanova, S.I.; Schreiber, B.E.; Ong, V.H.; Rosenberg, D.; Moinzadeh, P.; Coghlan, J.G.; Wells, A.U.; Denton, C.P. Prediction of Pulmonary Complications and Long-Term Survival in Systemic Sclerosis. Arthritis Rheumatol. 2014, 66, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, V.K.; Wirz, E.G.; Allanore, Y.; Rossbach, P.; Riemekasten, G.; Hachulla, E.; Distler, O.; Airò, P.; Carreira, P.E.; Balbir Gurman, A.; et al. Incidences and Risk Factors of Organ Manifestations in the Early Course of Systemic Sclerosis: A Longitudinal EUSTAR Study. PLoS ONE 2016, 11, e0163894. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, E.R.; Andréasson, K.; Smith, V. Systemic Sclerosis. Lancet 2023, 401, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.R.; Veeraraghavan, S.; Hansell, D.M.; Nikolakopolou, A.; Goh, N.S.L.; Nicholson, A.G.; Colby, T.V.; Denton, C.P.; Black, C.M.; du Bois, R.M.; et al. CT Features of Lung Disease in Patients with Systemic Sclerosis: Comparison with Idiopathic Pulmonary Fibrosis and Nonspecific Interstitial Pneumonia. Radiology 2004, 232, 560–567. [Google Scholar] [CrossRef]
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic Idiopathic Interstitial Lung Disease: The Molecular and Cellular Key Players. Int. J. Mol. Sci. 2021, 22, 8952. [Google Scholar] [CrossRef]
- Hochhegger, B.; Marchiori, E.; Zanon, M.; Rubin, A.S.; Fragomeni, R.; Altmayer, S.; Carvalho, C.R.R.; Baldi, B.G. Imaging in Idiopathic Pulmonary Fibrosis: Diagnosis and Mimics. Clinics 2019, 74, e225. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.K.; Ambrosini, R.D.; Kottmann, R.M.; Ritchlin, C.T.; Schwarz, E.M.; Rahimi, H. Reinterpreting Evidence of Rheumatoid Arthritis-Associated Interstitial Lung Disease to Understand Etiology. Curr. Rheumatol. Rev. 2019, 15, 277–289. [Google Scholar] [CrossRef]
- Cottin, V.; Brown, K.K. Interstitial Lung Disease Associated with Systemic Sclerosis (SSc-ILD). Respir. Res. 2019, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.L.; Fiorelli, R.F. Nonspecific Interstitial Pneumonia/Fibrosis. Histologic Features and Clinical Significance. Am. J. Surg. Pathol. 1994, 18, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, M.; Ichikawa, H.; Sato, S.; Tanaka, T.; Johkoh, T.; Kataoka, K.; Yamano, Y.; Kondoh, Y.; Nakamura, H.; Kawakami, A.; et al. Proposed Method of Histological Separation between Connective Tissue Disease-Associated Interstitial Pneumonia and Idiopathic Interstitial Pneumonias. PLoS ONE 2018, 13, e0206186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; du Bois, R.M. Histopathologic Subsets of Fibrosing Alveolitis in Patients with Systemic Sclerosis and Their Relationship to Outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef]
- Fischer, A.; Swigris, J.J.; Groshong, S.D.; Cool, C.D.; Sahin, H.; Lynch, D.A.; Curran-Everett, D.; Gillis, J.Z.; Meehan, R.T.; Brown, K.K. Clinically Significant Interstitial Lung Disease in Limited Scleroderma: Histopathology, Clinical Features, and Survival. Chest 2008, 134, 601–605. [Google Scholar] [CrossRef]
- Tanabe, N.; McDonough, J.E.; Vasilescu, D.M.; Ikezoe, K.; Verleden, S.E.; Xu, F.; Wuyts, W.A.; Vanaudenaerde, B.M.; Colby, T.V.; Hogg, J.C. Pathology of Idiopathic Pulmonary Fibrosis Assessed by a Combination of Microcomputed Tomography, Histology, and Immunohistochemistry. Am. J. Pathol. 2020, 190, 2427–2435. [Google Scholar] [CrossRef]
- Gallob, F.; Brcic, L.; Eidenhammer, S.; Rumpp, F.; Nerlich, A.; Popper, H. Senescence and Autophagy in Usual Interstitial Pneumonia of Different Etiology. Virchows Arch. 2021, 478, 497–506. [Google Scholar] [CrossRef]
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of Apoptotic and Antiapoptotic Markers in Epithelial Cells in Idiopathic Pulmonary Fibrosis. Chest 2005, 127, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, N.; Kuwano, K.; Maeyama, T.; Hagimoto, N.; Yoshimi, M.; Hamada, N.; Yamada, M.; Nakanishi, Y. The P53-Mdm2 Association in Epithelial Cells in Idiopathic Pulmonary Fibrosis and Non-Specific Interstitial Pneumonia. J. Clin. Pathol. 2005, 58, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Reed, J.C. Tumor Suppressor P53 Is a Direct Transcriptional Activator of the Human Bax Gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfei, M.; von der Beck, D.; Henneke, I.; Markart, P.; Ruppert, C.; Mahavadi, P.; Ghanim, B.; Klepetko, W.; Fink, L.; Meiners, S.; et al. Comparative Proteome Analysis of Lung Tissue from Patients with Idiopathic Pulmonary Fibrosis (IPF), Non-Specific Interstitial Pneumonia (NSIP) and Organ Donors. J. Proteom. 2013, 85, 109–128. [Google Scholar] [CrossRef] [Green Version]
- Markart, P.; Luboeinski, T.; Korfei, M.; Schmidt, R.; Wygrecka, M.; Mahavadi, P.; Mayer, K.; Wilhelm, J.; Seeger, W.; Guenther, A.; et al. Alveolar Oxidative Stress Is Associated with Elevated Levels of Nonenzymatic Low-Molecular-Weight Antioxidants in Patients with Different Forms of Chronic Fibrosing Interstitial Lung Diseases. Antioxid. Redox Signal. 2009, 11, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Ghislat, G.; Aguado, C.; Knecht, E. Annexin A5 Stimulates Autophagy and Inhibits Endocytosis. J. Cell Sci. 2012, 125, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margaritopoulos, G.A.; Tsitoura, E.; Tzanakis, N.; Spandidos, D.A.; Siafakas, N.M.; Sourvinos, G.; Antoniou, K.M. Self-Eating: Friend or Foe? The Emerging Role of Autophagy in Idiopathic Pulmonary Fibrosis. Biomed Res. Int. 2013, 2013, 420497. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular Senescence Mediates Fibrotic Pulmonary Disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [Green Version]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA Damage Triggers a Prolonged P53-Dependent G1 Arrest and Long-Term Induction of Cip1 in Normal Human Fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [Green Version]
- Gire, V.; Dulic, V. Senescence from G2 Arrest, Revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gil, J. Mitochondria and Senescence: New Actors for an Old Play. EMBO J. 2016, 35, 701–702. [Google Scholar] [CrossRef] [Green Version]
- Chilosi, M.; Poletti, V.; Murer, B.; Lestani, M.; Cancellieri, A.; Montagna, L.; Piccoli, P.; Cangi, G.; Semenzato, G.; Doglioni, C. Abnormal Re-Epithelialization and Lung Remodeling in Idiopathic Pulmonary Fibrosis: The Role of DeltaN-P63. Lab. Investig. 2002, 82, 1335–1345. [Google Scholar] [CrossRef] [Green Version]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated Epithelial Cell Senescence in IPF and the Inhibitory Role of SIRT6 in TGF-β-Induced Senescence of Human Bronchial Epithelial Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Devenish, R.J.; Klionsky, D.J. Autophagy: Mechanism and Physiological Relevance ‘Brewed’ from Yeast Studies. Front. Biosci. (Schol. Ed.) 2012, 4, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Deretic, V., Ed.; Methods in Molecular BiologyTM; Humana Press: Totowa, NJ, USA, 2008; pp. 77–88. ISBN 978-1-59745-157-4. [Google Scholar]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK Activation Protects Cells from Oxidative Stress-induced Senescence via Autophagic Flux Restoration and Intracellular NAD + Elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, I.; Muntean, L.; Crisan, T.; Rednic, V.; Sirbe, C.; Rednic, S. Novel Concepts in Systemic Sclerosis Pathogenesis: Role for MiRNAs. Biomedicines 2021, 9, 1471. [Google Scholar] [CrossRef] [PubMed]
- Farutin, V.; Kurtagic, E.; Pradines, J.R.; Capila, I.; Mayes, M.D.; Wu, M.; Manning, A.M.; Assassi, S. Multiomic Study of Skin, Peripheral Blood, and Serum: Is Serum Proteome a Reflection of Disease Process at the End-Organ Level in Systemic Sclerosis? Arthritis Res. Ther. 2021, 23, 259. [Google Scholar] [CrossRef] [PubMed]
- Sargent, J.L.; Whitfield, M.L. Capturing the Heterogeneity in Systemic Sclerosis with Genome-Wide Expression Profiling. Expert Rev. Clin. Immunol. 2011, 7, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Khanna, D. Systemic Sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SSc-ILD | Control Lung Donors (CLD) | Ref. | ||
|---|---|---|---|---|
| NSIP | UIP/IPF | |||
| Histologic Features | ||||
| Distribution of fibrosis | Uniform | Regional heterogeneity | Absent | [84] |
| Localization of fibrosis | Lower lobes | Lower lobes | Absent | [89] |
| Honeycomb change | Absent | Present | Absent | [85] |
| ILD | Control Lung Donors (CLD) | Ref | ||
|---|---|---|---|---|
| NSIP | UIP/IPF | |||
| Apoptosis | ||||
| p53 | Upregulated when compared with CLD Downregulated when compared with UIP/IPF | Upregulated | Homeostasis | [92,93] |
| Phosphorylated p53 | ||||
| Mdm2 | ||||
| p21 | ||||
| Bax | ||||
| p53-Mdm2 conjugate | - | Downregulated | Homeostasis | |
| HIPK2 kinase | Downregulated | - | Homeostasis | [94] |
| Autophagy | ||||
| LC3-II | Upregulated | Upregulated | Homeostasis | [90] |
| MAP1S | Upregulated | Upregulated | ||
| pAMPK | Upregulated | Upregulated | ||
| ANXA5 | Downregulated | Downregulated | [94,96] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spasovski, V.; Andjelkovic, M.; Parezanovic, M.; Komazec, J.; Ugrin, M.; Klaassen, K.; Stojiljkovic, M. The Role of Autophagy and Apoptosis in Affected Skin and Lungs in Patients with Systemic Sclerosis. Int. J. Mol. Sci. 2023, 24, 11212. https://doi.org/10.3390/ijms241311212
Spasovski V, Andjelkovic M, Parezanovic M, Komazec J, Ugrin M, Klaassen K, Stojiljkovic M. The Role of Autophagy and Apoptosis in Affected Skin and Lungs in Patients with Systemic Sclerosis. International Journal of Molecular Sciences. 2023; 24(13):11212. https://doi.org/10.3390/ijms241311212
Chicago/Turabian StyleSpasovski, Vesna, Marina Andjelkovic, Marina Parezanovic, Jovana Komazec, Milena Ugrin, Kristel Klaassen, and Maja Stojiljkovic. 2023. "The Role of Autophagy and Apoptosis in Affected Skin and Lungs in Patients with Systemic Sclerosis" International Journal of Molecular Sciences 24, no. 13: 11212. https://doi.org/10.3390/ijms241311212
APA StyleSpasovski, V., Andjelkovic, M., Parezanovic, M., Komazec, J., Ugrin, M., Klaassen, K., & Stojiljkovic, M. (2023). The Role of Autophagy and Apoptosis in Affected Skin and Lungs in Patients with Systemic Sclerosis. International Journal of Molecular Sciences, 24(13), 11212. https://doi.org/10.3390/ijms241311212