

Effects of a Peptide Derived from the Primary Sequence of a Kallikrein Inhibitor Isolated from Bauhinia bauhinioides (pep-BbKI) in an Asthma–COPD Overlap (ACO) Model
 , , , ,
, , , ,  , and
, and
Abstract

1. Introduction
2. Results
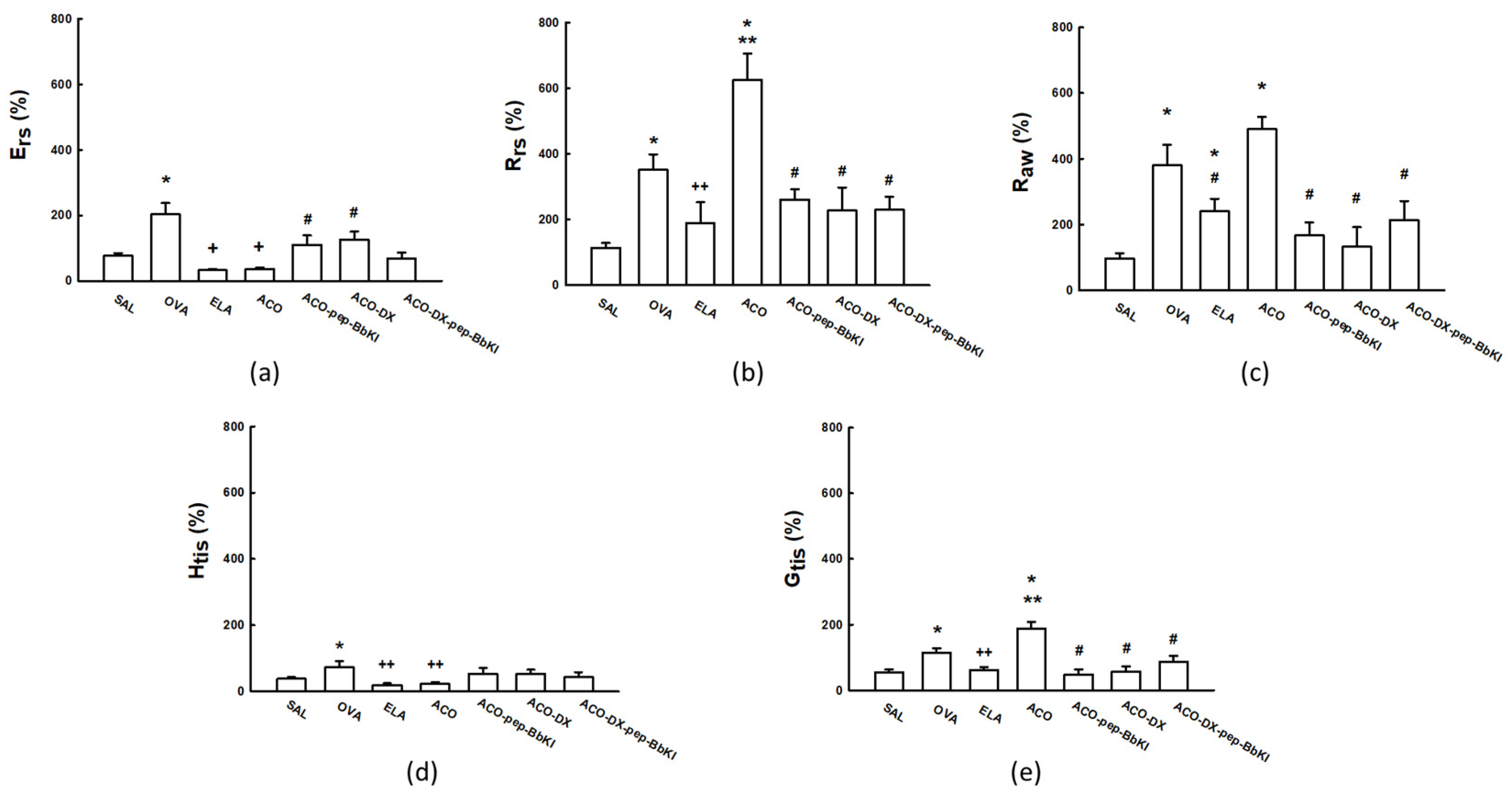
2.1. Hyperresponsiveness to Methacholine
2.2. Bronchoalveolar Lavage Fluid
2.3. Mean Linear Intercept (Lm)
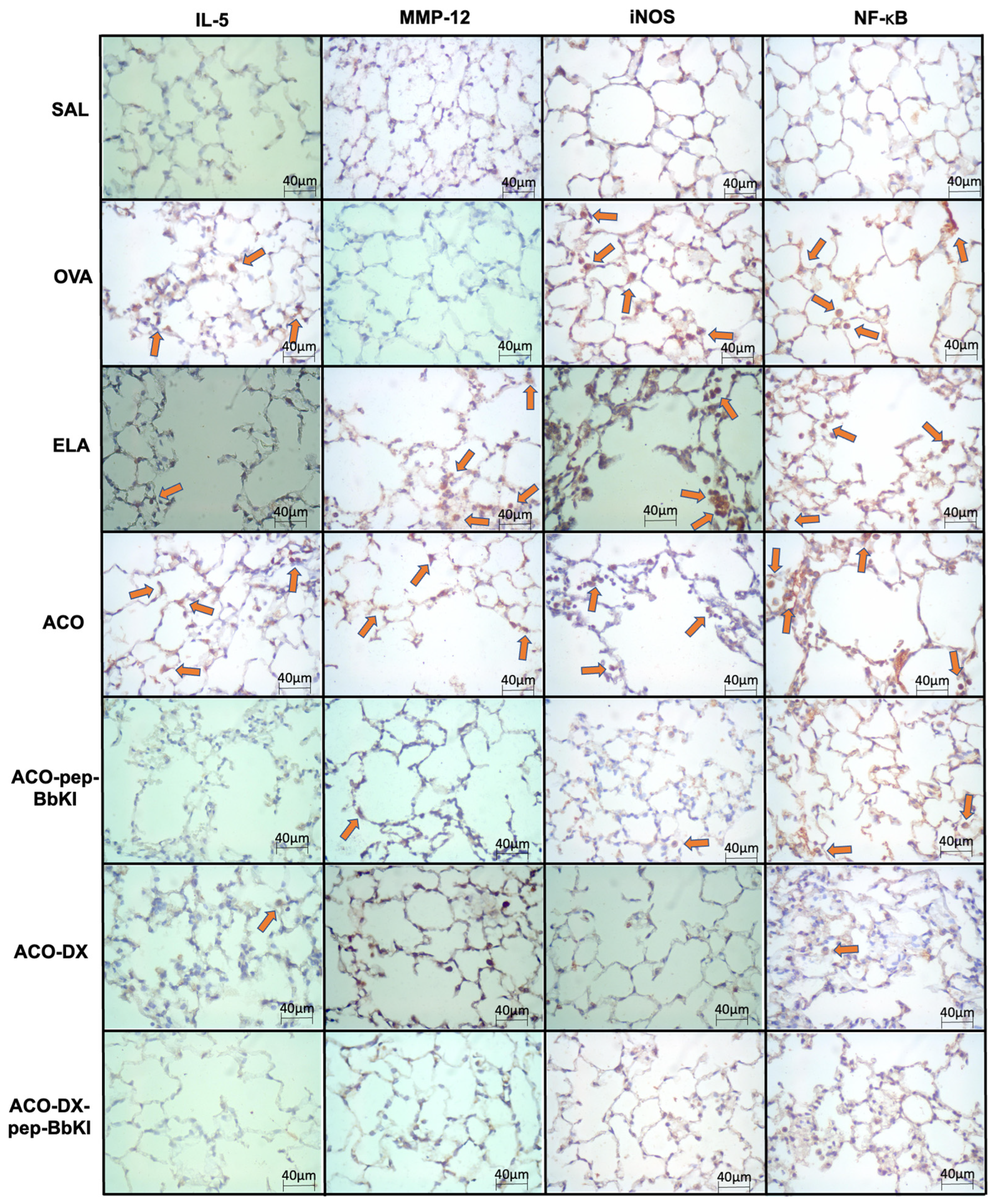
2.4. Lung Inflammation
2.5. Extracellular Matrix Remodeling
2.6. Oxidative Stress Response
2.7. Exhaled Nitric Oxide (eNO)
2.8. Signaling Pathway
2.9. Qualitative Analysis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Inhibitor Purification
4.3. Experimental Groups
4.4. Ovalbumin-Induced Asthma Mice Model
4.5. Elastase-Induced Emphysema Mice Model
4.6. Pep-BbKI and Dexamethasone Treatment Groups
4.7. Evaluation of Hyperresponsiveness to Methacholine and Determination of Exhaled Nitric Oxide
4.8. Bronchoalveolar Lavage Fluid (BALF) Analysis
4.9. Morphometric Studies
4.10. Immunohistochemistry
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACO | asthma-COPD overlap |
| ACO-DX | treated with dexamethasone |
| ACO-DX-pep-BbKI | treated with dexamethasone and inhibitor |
| ACO-pep-BbKI | treated with inhibitor |
| AS | alveolar septa |
| AW | airways |
| BALF | bronchoalveolar lavage fluid |
| BbCI | Bauhinia bauhinioides cruzipain proteinase inhibitor |
| BbkI | Bauhinia bauhinioides kallikrein inhibitor |
| BSA | bovine serum albumin |
| CD4+ | cluster of differentation 4 |
| CD8+ | cluster of differentation 8 |
| CNPq | National Council for Scientific and Technological Development |
| COPD | chronic obstructive pulmonary disease |
| CrataBL | Crataeva tapia Bark Lectin |
| DAB | diaminobenzidine chromogen |
| EcTI | Enterolobium contortisiliquum |
| ELA | elastase protocol |
| eNO | exhaled nitric oxide |
| %Ers | respiratory system elastance |
| FAPESP | research support foundation of the State of São Paulo |
| FEV1 | forced expiratory volume in first second |
| GINA | Global Initiative of Asthma |
| GOLD | Global Initiative for Chronic Obstructive Lung Disease |
| %Gtis | tissue resistance system |
| %Htis | tissue elastance system |
| IFN-γ | interferon gamma |
| IgE | immunoglobulin E |
| IL-1β | interleukin 1 Beta |
| IL-4 | interleukin 4 |
| IL-5 | interleukin 5 |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| IL-13 | interleukin 13 |
| IL-17 | interleukin 17 |
| iNOS | inducible nitric oxide synthase |
| IP | intraperitoneal |
| Lm | linear mean intercept |
| LIM-20 | Laboratory of Experimental Therapy I |
| MMP-9 | metalloproteinase 9 |
| MMP-12 | metalloproteinase 12 |
| NF-κB | nuclear factor kappa-B |
| OVA | ovalbumin protocol |
| PBS | Phosphate-buffered Saline |
| Pep-BbKI | Bauhinia bauhinioides |
| PKa | human plasma kallikrein |
| ppb | parts per billion |
| PPE | porcine pancreatic elastase |
| %Raw | airway resistance system |
| rBmTI-A | Boophilus microplus trypsin inhibitor |
| %Rrs | respiratory system resistance |
| SAL | saline protocol |
| SAL-pep-BbKI | saline group treated with inhibitor |
| TGF-β | transforming growth factor beta |
| Th2 | T-helper cells 2 |
| Th17 | T-helper cells 17 |
| TNF-α | tumor necrosis factor alpha |
References
- Viegi, G.; Pistelli, F.; Sherrill, D.L.; Maio, S.; Baldacci, S.; Carrozzi, L. Definition, epidemiology and natural history of COPD. Eur. Respir. J. 2007, 30, 993–1013. [Google Scholar] [CrossRef]
- Putcha, N.; Wise, R.A. Asthma-Chronic Obstructive Pulmonary Disease Overlap Syndrome: Nothing New Under the Sun. Immunol. Allergy Clin. N. Am. 2016, 36, 515–528. [Google Scholar] [CrossRef]
- Global Initiative for Asthma (GINA). Global Strategy for Asthma Management and Prevention Program. 2022. Available online: http://www.ginaasthma.org (accessed on 28 March 2022).
- Ling, M.F.; Luster, A.D. Allergen-Specific CD4+ T Cells in Human Asthma. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S1), S25–S30. [Google Scholar] [CrossRef]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in a cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 26. [Google Scholar] [CrossRef]
- Pavord, I.D.; Brightling, C.E.; Woltmann, G.; Wardlaw, A.J. Non-eosinophilic corticosteroid unresponsive asthma. Lancet 1999, 353, 2213–2214. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.; Krysko, D.V.; Vandenabeele, P.; de Vries, M.; van Oosterhout, A.J.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L377–L386. [Google Scholar] [CrossRef]
- Gafar, F.; Boudewijn, I.M.; Cox, C.A.; Vonk, J.M.; Schokker, S.; Lexmond, A.J.; Frijlink, H.W.; Hagedoorn, P.; Postma, D.S.; van den Berge, M. Predictors of clinical response to extrafine and non-extrafine particle inhaled corticosteroids in smokers and ex-smokers with asthma. Respir. Res. 2018, 19, 256. [Google Scholar] [CrossRef]
- Maselli, D.J.; Hardin, M.; Christenson, S.A.; Hanania, N.A.; Hersh, C.P.; Adams, S.G.; Anzueto, A.; Peters, J.I.; Han, M.K.; Martinez, F.J. Clinical Approach to the Therapy of Asthma-COPD Overlap. Chest 2019, 155, 168–177. [Google Scholar] [CrossRef]
- Wu, W.; Bang, S.; Bleecker, E.R.; Castro, M.; Denlinger, L.; Erzurum, S.C.; Fahy, J.V.; Fitzpatrick, A.M.; Gaston, B.M.; Hastie, A.T.; et al. Multiview Cluster Analysis Identifies Variable Corticosteroid Response Phenotypes in Severe Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M. Seed storage proteins: The enzyme inhibitors. Methods Plant Biochem. 1991, 5, 259–305. [Google Scholar]
- Barnes, P.J.; Stockley, R.A. COPD: Current therapeutic interventions and future approaches. Eur. Respir. J. 2005, 25, 1084–1106. [Google Scholar] [CrossRef] [PubMed]
- Bonturi, C.R.; Silva Teixeira, A.B.; Rocha, V.M.; Valente, P.F.; Oliveira, J.R.; Filho, C.M.B.; Fátima Correia Batista, I.; Oliva, M.L.V. Plant Kunitz Inhibitors and Their Interaction with Proteases: Current and Potential Pharmacological Targets. Int. J. Mol. Sci. 2022, 23, 4742. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.L.; Sampaio, M.U. Action of plant proteinase inhibitors on enzymes of physiopathological importance. An. Acad. Bras. Cienc. 2009, 81, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Mendes, C.R.; Juliano, M.A.; Chagas, J.R.; Rosa, J.C.; Greene, L.J.; Sampaio, M.U.; Sampaio, C.A. Characterization of a tissue kallikrein inhibitor isolated from Bauhinia bauhinioides seeds: Inhibition of the hydrolysis of kininogen related substrates. Immunopharmacology 1999, 45, 163–169. [Google Scholar] [CrossRef]
- Araújo, A.P.; Hansen, D.; Vieira, D.F.; Oliveira, C.; Santana, L.A.; Beltramini, L.M.; Sampaio, C.A.; Sampaio, M.U.; Oliva, M.L. Kunitz-type Bauhinia bauhinioides inhibitors devoid of disulfide bridges: Isolation of the cDNAs, heterologous expression and structural studies. Biol. Chem. 2005, 386, 561–568. [Google Scholar] [CrossRef]
- Brito, M.V.; de Oliveira, C.; Salu, B.R.; Andrade, S.A.; Malloy, P.M.; Sato, A.C.; Vicente, C.P.; Sampaio, M.U.; Maffei, F.H.; Oliva, M.L. The Kallikrein Inhibitor from Bauhinia bauhinioides (BbKI) shows antithrombotic properties in venous and arterial thrombosis models. Thromb. Res. 2014, 133, 945–951. [Google Scholar] [CrossRef]
- Oliva, M.L.; Mendes, C.R.; Santomauro-Vaz, E.M.; Juliano, M.A.; Mentele, R.; Auerswald, E.A.; Sampaio, M.U.; Sampaio, C.A. Bauhinia bauhinioides plasma kallikrein inhibitor: Interaction with synthetic peptides and fluorogenic peptide substrates related to the reactive site sequence. Curr. Med. Chem. 2001, 8, 977–984. [Google Scholar] [CrossRef]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Reis, R.; Theodoro-Junior, O.A.; Oliveira, B.T.M.; Oliva, L.V.; Toledo-Arruda, A.C.; Bonturi, C.R.; Brito, M.V.; Lopes, F.D.T.Q.S.; Prado, C.M.; Florencio, A.C.; et al. Plant Proteinase Inhibitor BbCI Modulates Lung Inflammatory Responses and Mechanic and Remodeling Alterations Induced by Elastase in Mice. Biomed. Res. Int. 2017, 2017, 8287125. [Google Scholar] [CrossRef]
- Bortolozzo, A.S.S.; Rodrigues, A.P.D.; Arantes-Costa, F.M.; Saraiva-Romanholo, B.M.; de Souza, F.C.R.; Brüggemann, T.R.; de Brito, M.V.; Ferreira, R.D.S.; Correia, M.T.D.S.; Paiva, P.M.G.; et al. The Plant Proteinase Inhibitor CrataBL Plays a Role in Controlling Asthma Response in Mice. Biomed. Res. Int. 2018, 2018, 9274817. [Google Scholar] [CrossRef]
- Florencio, A.C.; de Almeida, R.S.; Arantes-Costa, F.M.; Saraiva-Romanholo, B.M.; Duran, A.F.; Sasaki, S.D.; Martins, M.A.; Lopes, F.D.T.Q.S.; Tibério, I.F.L.C.; Leick, E.A. Effects of the serine protease inhibitor rBmTI-A in an experimental mouse model of chronic allergic pulmonary inflammation. Sci. Rep. 2019, 9, 12624. [Google Scholar] [CrossRef]
- Martins-Olivera, B.T.; Almeida-Reis, R.; Theodoro-Júnior, O.A.; Oliva, L.V.; Neto Dos Santos Nunes, N.; Olivo, C.R.; Vilela de Brito, M.; Prado, C.M.; Leick, E.A.; Martins, M.d.A.; et al. The Plant-Derived Bauhinia bauhinioides Kallikrein Proteinase Inhibitor (rBbKI) Attenuates Elastase-Induced Emphysema in Mice. Mediat. Inflamm. 2016, 2016, 5346574. [Google Scholar] [CrossRef] [PubMed]
- Theodoro-Júnior, O.A.; Righetti, R.F.; Almeida-Reis, R.; Martins-Oliveira, B.T.; Oliva, L.V.; Prado, C.M.; Saraiva-Romanholo, B.M.; Leick, E.A.; Pinheiro, N.M.; Lobo, Y.A.; et al. A Plant Proteinase Inhibitor from Enterolobium contortisiliquum Attenuates Pulmonary Mechanics, Inflammation and Remodeling Induced by Elastase in Mice. Int. J. Mol. Sci. 2017, 18, 403. [Google Scholar] [CrossRef]
- Oliva, L.V.; Almeida-Reis, R.; Theodoro-Junior, O.; Oliveira, B.M.; Leick, E.A.; Prado, C.M.; Brito, M.V.; dos Santos Correia, M.T.; Paiva, P.M.G.; Martins, M.A.; et al. A plant proteinase inhibitor from Crataeva tapia (CrataBL) attenuates elastase-induced pulmonary inflammatory, remodeling, and mechanical alterations in mice. Process Biochem. 2015, 50, 1958–1965. [Google Scholar] [CrossRef]
- Rodrigues, A.P.D.; Bortolozzo, A.S.S.; Arantes-Costa, F.M.; Saraiva-Romanholo, B.M.; de Souza, F.C.R.; Brüggemann, T.R.; Santana, F.P.R.; de Brito, M.V.; Bonturi, C.R.; Nunes, N.N.D.S.; et al. A plant proteinase inhibitor from Enterolobium contortisiliquum attenuates airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Histol. Histopathol. 2019, 34, 537–552. [Google Scholar] [CrossRef]
- Ikeda, G.; Miyahara, N.; Koga, H.; Fuchimoto, Y.; Waseda, K.; Kurimoto, E.; Taniguchi, A.; Tanimoto, Y.; Kataoka, M.; Tanimoto, M.; et al. Effect of a cysteinyl leukotriene receptor antagonist on experimental emphysema and asthma combined with emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 50, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ingenito, E.P.; Arold, S.P.; Parameswaran, H.; Tgavalekos, N.T.; Lutchen, K.R.; Suki, B. Tissue heterogeneity in the mouse lung: Effects of elastase treatment. J. Appl. Physiol. 2004, 97, 204–212. [Google Scholar] [CrossRef]
- Ito, S.; Ingenito, E.P.; Brewer, K.K.; Black, L.D.; Parameswaran, H.; Lutchen, K.R.; Suki, B. Mechanics, nonlinearity, and failure strength of lung tissue in a mouse model of emphysema: Possible role of collagen remodeling. J. Appl. Physiol. 2005, 98, 503–511. [Google Scholar] [CrossRef]
- Scuri, M.; Forteza, R.; Lauredo, I.; Sabater, J.R.; Botvinnikova, Y.; Allegra, L.; Abraham, W.M. Inhaled porcine pancreatic elastase causes bronchoconstriction via a bradykinin-mediated mechanism. J. Appl. Physiol. 2000, 89, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Neuhof, C.; Oliva, M.L.; Maybauer, D.; Maybauer, M.; de Oliveira, C.; Sampaio, M.U.; Sampaio, C.A.; Neuhof, H. Effect of plant Kunitz inhibitors from Bauhinia bauhinioides and Bauhinia rufa on pulmonary edema caused by activated neutrophils. Biol. Chem. 2003, 384, 939–944. [Google Scholar] [CrossRef]
- Lourenço, J.D.; Neves, L.P.; Olivo, C.R.; Duran, A.; Almeida, F.M.; Arantes, P.M.; Prado, C.M.; Leick, E.A.; Tanaka, A.S.; Martins, M.A.; et al. A treatment with a protease inhibitor recombinant from the cattle tick (Rhipicephalus Boophilus microplus) ameliorates emphysema in mice. PLoS ONE 2014, 9, e98216. [Google Scholar] [CrossRef]
- Global Strategy for Diagnosis Management, and Prevention of COPD, Updated 2022. Available online: https://goldcopd.org/2022-gold-reports-2/ (accessed on 20 May 2022).
- Raundhal, M.; Morse, C.; Khare, A.; Oriss, T.B.; Milosevic, J.; Trudeau, J.; Huff, R.; Pilewski, J.; Holguin, F.; Kolls, J.; et al. High IFN-γ and low SLPI mark severe asthma in mice and humans. J. Clin. Investig. 2015, 125, 3037–3050. [Google Scholar] [CrossRef]
- Camargo, L.D.N.; Righetti, R.F.; Aristóteles, L.R.C.R.B.; Dos Santos, T.M.; de Souza, F.C.R.; Fukuzaki, S.; Cruz, M.M.; Alonso-Vale, M.I.C.; Saraiva-Romanholo, B.M.; Prado, C.M.; et al. Effects of Anti-IL-17 on Inflammation, Remodeling, and Oxidative Stress in an Experimental Model of Asthma Exacerbated by LPS. Front. Immunol. 2018, 8, 1835. [Google Scholar] [CrossRef] [PubMed]
- Possa, S.S.; Charafeddine, H.T.; Righetti, R.F.; da Silva, P.A.; Almeida-Reis, R.; Saraiva-Romanholo, B.M.; Perini, A.; Prado, C.M.; Leick-Maldonado, E.A.; Martins, M.A.; et al. Rho-kinase inhibition attenuates airway responsiveness, inflammation, matrix remodeling, and oxidative stress activation induced by chronic inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L939–L952. [Google Scholar] [CrossRef] [PubMed]
- Mahmutovic Persson, I.; Menzel, M.; Ramu, S.; Cerps, S.; Akbarshahi, H.; Uller, L. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir. Res. 2018, 19, 16. [Google Scholar] [CrossRef]
- Hamid, Q.; Tulic, M. Immunobiology of asthma. Annu. Rev. Physiol. 2009, 71, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, T.M.; Righetti, R.F.; Camargo, L.D.N.; Saraiva-Romanholo, B.M.; Aristoteles, L.R.C.R.B.; de Souza, F.C.R.; Fukuzaki, S.; Alonso-Vale, M.I.C.; Cruz, M.M.; Prado, C.M.; et al. Effect of Anti-IL17 Antibody Treatment Alone and in Combination with Rho-Kinase Inhibitor in a Murine Model of Asthma. Front. Physiol. 2018, 9, 1183. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef]
- Parulekar, A.D.; Diamant, Z.; Hanania, N.A. Role of biologics targeting type 2 airway inflammation in asthma: What have we learned so far? Curr. Opin. Pulm. Med. 2017, 23, 3–11. [Google Scholar] [CrossRef]
- Park, H.Y.; Lee, H.; Koh, W.J.; Kim, S.; Jeong, I.; Koo, H.K.; Kim, T.H.; Kim, J.W.; Kim, W.J.; Oh, Y.M.; et al. Association of blood eosinophils and plasma periostin with FEV1 response after 3-month inhaled corticosteroid and long-acting beta2-agonist treatment in stable COPD patients. Int. J. Chron. Obstruct. Pulmon. Dis. 2015, 11, 23–30. [Google Scholar] [CrossRef]
- Ponce-Gallegos, M.A.; Ramírez-Venegas, A.; Falfán-Valencia, R. Th17 profile in COPD exacerbations. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, G.J.; Ten Hacken, N.H.; Hoffmann, R.F.; van Oosterhout, A.J.; Heijink, I.H. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur. Respir. J. 2012, 39, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Fukuzaki, S.; Righetti, R.F.; Santos, T.M.D.; Camargo, L.D.N.; Aristóteles, L.R.C.R.B.; Souza, F.C.R.; Garrido, A.C.; Saraiva-Romanholo, B.M.; Leick, E.A.; Prado, C.M.; et al. Preventive and therapeutic effect of anti-IL-17 in an experimental model of elastase-induced lung injury in C57Bl6 mice. Am. J. Physiol. Cell Physiol. 2021, 320, C341–C354. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.M.; Leick-Maldonado, E.A.; Arata, V.; Kasahara, D.I.; Martins, M.A.; Tibério, I.F. Neurokinins and inflammatory cell iNOS expression in guinea pigs with chronic allergic airway inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L741–L748. [Google Scholar] [CrossRef]
- Kharitonov, S.A.; Yates, D.H.; Barnes, P.J. Inhaled glucocorticoids decrease nitric oxide in exhaled air of asthmatic patients. Am J. Respir. Crit. Care Med. 1996, 153, 454–457. [Google Scholar] [CrossRef]
- Eynott, P.R.; Groneberg, D.A.; Caramori, G.; Adcock, I.M.; Donnelly, L.E.; Kharitonov, S.; Barnes, P.J.; Chung, K.F. Role of nitric oxide in allergic inflammation and bronchial hyperresponsiveness. Eur. J. Pharmacol. 2002, 452, 123–133. [Google Scholar] [CrossRef]
- Suki, B.; Lutchen, K.R.; Ingenito, E.P. On the progressive nature of emphysema: Roles of proteases, inflammation, and mechanical forces. Am. J. Respir. Crit. Care Med. 2003, 168, 516–521. [Google Scholar] [CrossRef]
- Rico-Rosillo, G.; Vega-Robledo, G.B. The involvement of NF-κB Transcription factor in asthma. Rev. Alerg. Mex. 2011, 58, 107–111. [Google Scholar]
- Aristoteles, L.R.; Righetti, R.F.; Pinheiro, N.M.; Franco, R.B.; Starling, C.M.; da Silva, J.C.P.; Pigati, P.A.; Caperuto, L.C.; Prado, C.M.; Dolhnikoff, M.; et al. Modulation of the oscillatory mechanics of lung tissue and the oxidative stress response induced by arginase inhibition in a chronic allergic inflammation model. BMC Pulm. Med. 2013, 13, 52. [Google Scholar] [CrossRef]
- Pantano, C.; Ather, J.L.; Alcorn, J.F.; Poynter, M.E.; Brown, A.L.; Guala, A.S.; Beuschel, S.L.; Allen, G.B.; Whittaker, L.A.; Bevelander, M.; et al. Nuclear factor-kappaB activation in airway epithelium induces inflammation and hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2008, 177, 959–969. [Google Scholar] [CrossRef]
- Sumikawa, J.T.; Brito, M.V.; Macedo, M.L.; Uchoa, A.F.; Miranda, A.; Araujo, A.P.; Silva-Lucca, R.A.; Sampaio, M.U.; Oliva, M.L. The defensive functions of plant inhibitors are not restricted to insect enzyme inhibition. Phytochemistry 2010, 71, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Toledo, A.C.; Sakoda, C.P.; Perini, A.; Pinheiro, N.M.; Magalhães, R.M.; Grecco, S.; Tibério, I.F.; Câmara, N.O.; Martins, M.A.; Lago, J.H.; et al. Flavonone treatment reverses airway inflammation and remodelling in an asthma murine model. Br. J. Pharmacol. 2013, 168, 1736–1749. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.H.; Irvin, C.G. Measuring lung function in mice: The phenotyping uncertainty principle. J. Appl. Physiol. 2003, 94, 1297–1306. [Google Scholar] [CrossRef]
- Biselli, P.J.C.; Kohler, J.B.; Righetti, R.; Tibério, I.F.L.C.; Martins, M.A.; Lopes, F.D.T.Q.S. Analysis of respiratory mechanics in animal models: Its use in understanding lung behavior in emphysema and asthma. Drug Discov. Today Dis. Model. 2019, 29–30, 11–17. [Google Scholar] [CrossRef]
- Weibel, E.R. The challenge of measuring lung structure. On the “Standards for the Quantitative Assessment of Lung Structure”. Nihon Kokyuki Gakkai Zasshi 2010, 48, 637–643. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflammatory Markers (Cells/104 µm2) | SAL | OVA | ELA | ACO | ACO-pep-BbKI | ACO-DX | ACO-DX-pep-BbKI |
|---|---|---|---|---|---|---|---|
| IL-1β—Airways | 0.4 ± 0.1 | 4.2 ± 0.4 * | 1.2 ± 0.2 ++ | 6.0 ± 0.5 */** | 2.7 ± 0.3 */#/$ | 1.4 ± 0.3 */# | 2.2 ± 0.2 */# |
| IL-1β—Alveolar septa | 0.2 ± 0.1 | 2.6 ± 0.3 * | 0.7 ± 0.2 ++ | 4.0 ± 0.3 */** | 0.9 ± 0.2 */# | 0.5 ± 0.1 # | 1.3 ± 0.2 */# |
| IL-4—Airways | 1.5 ± 0.5 | 11.3 ± 1.3 * | 4.1 ± 1.2 #/++ | 12.2 ± 2.3 * | 11.9 ± 1.7 * | 7.9 ± 1.2 * | 8.7 ± 1.2 * |
| IL-4—Alveolar septa | 1.7 ± 0.3 | 10.4 ± 0.6 * | 4.7 ± 0.7 */++ | 7.9 ± 0.7 */** | 8.8 ± 0.7 * | 6.5 ± 0.5 * | 6.5 ± 0.7 * |
| IL-5—Airways | 1.7 ± 0.2 | 11.3 ± 0.7 * | 6.5 ± 0.5 */++ | 8.5 ± 0.6 */** | 2.4 ± 0.4 #/$ | 6.6 ± 0.5 */# | 2.7 ± 0.5 #/$ |
| IL-5—Alveolar septa | 1.2 ± 0.2 | 4.4 ± 0.3 * | 2.9 ± 0.6 */++ | 8.0 ± 0.4 */** | 1.9 ± 0.3 # | 2.0 ± 0.2 # | 1.7 ± 0.2 # |
| IL-6—Airways | 0.7 ± 0.4 | 8.3 ± 0.5 * | 5.5 ± 0.4 */#/++ | 7.4 ± 0.6 * | 2.2 ± 0.2 */# | 1.9 ± 0.3 */# | 1.8 ± 0.2 */# |
| IL-6—Alveolar septa | 0.4 ± 0.1 | 6.6 ± 0.4 * | 7.2 ± 0.5 * | 12.3 ± 1.1 */** | 1.9 ± 0.2 */# | 1.1 ± 0.2 #/$$ | 1.0 ± 0.2 #/$$ |
| IL-10—Airways | 2.1 ± 0.2 | 3.3 ± 0.2 * | 4.1 ± 0.3 * | 5.2 ± 0.4 */++ | 2.8 ± 0.3#/$ | 4.1 ± 0.3 * | 2.3 ± 0.2 #/$ |
| IL-10—Alveolar septa | 3.2 ± 0.3 | 4.5 ± 0.3 * | 5.5 ± 0.4 * | 6.6 ± 0.4 */++ | 2.3 ± 0.2# | 3.6 ± 0.3 #/$$ | 3.3 ± 0.3 #/$$ |
| IL-13—Airways | 1.7 ± 0.2 | 10.4 ± 0.7 * | 7.9 ± 0.7 */#/++ | 11.6 ± 0.6 * | 4.7 ± 0.4 */# | 5.8 ± 0.4 */# | 4.3 ± 0.3 */#/$ |
| IL-13—Alveolar septa | 2.6 ± 0.4 | 9.2 ± 0.6 * | 6.7 ± 0.6 */++ | 16.9 ± 1.5 */** | 5.2 ± 0.4 */# | 4.2 ± 0.3 # | 6.0 ± 0.9 */# |
| IL-17—Airways | 2.1 ± 0.2 | 5.6 ± 0.3 * | 6.4 ± 0.3 * | 7.3 ± 0.4 + | 3.1 ± 0.2 # | 4.0 ± 0.3 */# | 3.7 ± 0.2 */# |
| IL-17—Alveolar septa | 1.5 ± 0.2 | 6.5 ± 0.3 * | 8.1 ± 0.4+ | 8.4 ± 0.6 + | 2.7 ± 0.2 #/$ | 4.9 ± 0.3 */# | 3.2 ± 0.6 */#/$ |
| IFN-γ—Airways | 0.7 ± 0.2 | 2.1 ± 0.2 | 3.3 ± 0.4 * | 6.2 ± 0.7 */** | 1.1 ± 0.2 #/$ | 0.3 ± 0.1 # | 1.9 ± 0.3 */#/$ |
| IFN-γ—Alveolar septa | 0.7 ± 0.2 | 2.9 ± 0.3 * | 2.4 ± 0.3 * | 3.2 ± 0.3 * | 1.2 ± 0.2 # | 0.6 ± 0.2 # | 0.9 ± 0.2 # |
| TNF-α—Airways | 1.7 ± 0.4 | 6.2 ± 0.7 * | 5.3 ± 0.7 * | 5.0 ± 0.7 * | 2.8 ± 0.4 | 5.2 ± 0.7 */$$ | 4.9 ± 0.7 */$$ |
| TNF-α-Alveolar septa | 1.3 ± 0.2 | 5.5 ± 0.4 */# | 6.2 ± 0.6 * | 7.7 ± 0.5 * | 2.1 ± 0.3 # | 4.5 ± 0.4 */#/$$ | 4.8 ± 0.4 */#/$$ |
| Remodeling Markers | SAL | OVA | ELA | ACO | ACO-pep-BbKI | ACO-DX | ACO-DX-pep-BbKI |
|---|---|---|---|---|---|---|---|
| MMP-9—Airways (cells/104 µm2) | 0.2 ± 0.03 | 3.0 ± 0.1 * | 5.3 ± 0.2 * | 10.2 ± 0.3 */** | 1.8 ± 0.08 */# | 2.1 ± 0.08 */# | 1.7 ± 0.1 */#/$ |
| MMP-9—Alveolar septa (cells/104 µm2) | 0.5 ± 0.1 | 2.1 ± 0.08 * | 4.4 ± 0.1 * | 8.7 ± 0.3 */** | 1.9 ± 0.1 */# | 2.3 ± 0.1 */# | 1.5 ± 0.1 */#/$ |
| MMP-12—Airways (cells/104 µm2) | 1.3 ± 0.2 | 1.9 ± 0.2 | 2.1 ± 0.4 | 3.5 ± 0.4 */** | 1.8 ± 0.2 # | 0.9 ± 0.1 #/$$ | 1.0 ± 0.2 #/$$ |
| MMP-12—Alveolar septa (cells/104 µm2) | 0.4 ± 0.1 | 0.2 ± 0.08 | 2.7 ± 0.4 * | 5.7 ± 0.5 */** | 2.5 ± 0.3 */# | 2.2 ± 0.3 */# | 0.9 ± 0.2 #/$/$$ |
| TGF-β—Airways (cells/104 µm2) | 0.8 ± 0.3 | 5.5 ± 1.1 * | 2.7 ± 0.4 # | 7.0 ± 0.9 * | 3.9 ± 0.4 */# | 3.0 ± 0.4 */# | 1.6 ± 0.2 #/$$ |
| TGF-β—Alveolar septa (cells/104 µm2) | 0.1 ± 0.07 | 4.8 ± 0.3 * | 3.6 ± 0.4 * | 6.4 ± 0.5 */** | 2.3 ± 0.3 */#/$ | 0.7 ± 0.2 # | 1.9 ± 0.2 */#/$ |
| Collagen fibers—Airways (%) | 1.6 ± 0.4 | 12.5 ± 0.4 * | 0.3 ± 0.1 ++/# | 12.0 ± 1.7 * | 1.8 ± 0.4 # | 1.3 ± 0.2 # | 3.0 ± 2.4 # |
| Collagen fibers—Alveolar septa (%) | 2.4 ± 0.2 | 11.3 ± 0.3 * | 3.9 ± 0.5 ++ | 9.0 ± 0.8 */** | 2.9 ± 0.3 #/$ | 5.7 ± 0.7 */# | 2.5 ± 0.2 #/$ |
| Marker | Specifications Primary Antibody | Dilution | Secondary Antibody | Specifications Secondary Antibody |
|---|---|---|---|---|
| IL-1β | SC-52012, L: A0719; Santa Cruz Biotechnology, CA, USA. | 1:50 | anti-mouse | L: ZG0715, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| IL-4 | SC-53084, L: J1518; Santa Cruz Biotechnology, CA, USA. | 1:8000 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| IL-5 | SC-398334, L: F1617; Santa Cruz Biotechnology, CA, USA. | 1:300 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| IL-6 | LS-C746886, L: 144178; LSBio, WA, USA. | 1:200 | anti-rabbit | L: ZF0103, Vector; Vectastin Elite ABC Kit (Rabbit IgG), CA, USA. |
| IL-10 | SC-8438; Santa Cruz Biotechnology, CA, USA. | 1:50 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| IL-13 | SC-393365, L:G1715; Santa Cruz Biotechnology, CA, USA. | 1:8000 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| IL-17 | SC-7927, L:A3113; Santa Cruz Biotechnology, CA, USA. | 1:100 | anti-rabbit | L: ZF0103, Vector; Vectastin Elite ABC Kit (Rabbit IgG), CA, USA. |
| IFN-γ | SC-8308, L:B2811; Santa Cruz Biotechnology, CA, USA. | 1:100 | anti-rabbit | L: ZF0103, Vector; Vectastin Elite ABC Kit (Rabbit IgG), CA, USA. |
| TNF-α | SC-52746, L: J2418; Santa Cruz Biotechnology, CA, USA. | 1:5000 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| MMP-9 | SC-393859, L: 6118; Santa Cruz Biotechnology, CA, USA. | 1:800 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| MMP-12 | SC-30072, L: B1910; Santa Cruz Biotechnology, CA, USA. | 1:400 | anti-rabbit | L: ZF0103, Vector; Vectastin Elite ABC Kit (Rabbit IgG), CA, USA. |
| TGF-β | SC-130348, L: A0219; Santa Cruz Biotechnology, CA, USA. | 1:700 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
| iNOS | RB-9242-P, L: 9242P709C; Thermo Fisher Scientific, UK. | 1:150 | anti-rabbit | L: ZF0103, Vector; Vectastin Elite ABC Kit (Rabbit IgG), CA, USA. |
| NF-κB | SC-8008, L: B1119; Santa Cruz Biotechnology, CA, USA. | 1:700 | anti-mouse | L: ZF0206, Vector; Vectastain Elite ABC Kit Peroxidase (Mouse IgG), CA, USA. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, L.L.S.d.; Barbosa, J.A.S.; João, J.M.L.G.; Fukuzaki, S.; Camargo, L.d.N.; dos Santos, T.M.; Campos, E.C.d.; Costa, A.S.; Saraiva-Romanholo, B.M.; Bezerra, S.K.M.; et al. Effects of a Peptide Derived from the Primary Sequence of a Kallikrein Inhibitor Isolated from Bauhinia bauhinioides (pep-BbKI) in an Asthma–COPD Overlap (ACO) Model. Int. J. Mol. Sci. 2023, 24, 11261. https://doi.org/10.3390/ijms241411261
Silva LLSd, Barbosa JAS, João JMLG, Fukuzaki S, Camargo LdN, dos Santos TM, Campos ECd, Costa AS, Saraiva-Romanholo BM, Bezerra SKM, et al. Effects of a Peptide Derived from the Primary Sequence of a Kallikrein Inhibitor Isolated from Bauhinia bauhinioides (pep-BbKI) in an Asthma–COPD Overlap (ACO) Model. International Journal of Molecular Sciences. 2023; 24(14):11261. https://doi.org/10.3390/ijms241411261
Chicago/Turabian StyleSilva, Luana Laura Sales da, Jéssica Anastácia Silva Barbosa, Juliana Morelli Lopes Gonçalves João, Silvia Fukuzaki, Leandro do Nascimento Camargo, Tabata Maruyama dos Santos, Elaine Cristina de Campos, Arthur Silva Costa, Beatriz Mangueira Saraiva-Romanholo, Suellen Karoline Moreira Bezerra, and et al. 2023. "Effects of a Peptide Derived from the Primary Sequence of a Kallikrein Inhibitor Isolated from Bauhinia bauhinioides (pep-BbKI) in an Asthma–COPD Overlap (ACO) Model" International Journal of Molecular Sciences 24, no. 14: 11261. https://doi.org/10.3390/ijms241411261
APA StyleSilva, L. L. S. d., Barbosa, J. A. S., João, J. M. L. G., Fukuzaki, S., Camargo, L. d. N., dos Santos, T. M., Campos, E. C. d., Costa, A. S., Saraiva-Romanholo, B. M., Bezerra, S. K. M., Lopes, F. T. Q. d. S., Bonturi, C. R., Oliva, M. L. V., Leick, E. A., Righetti, R. F., & Tibério, I. d. F. L. C. (2023). Effects of a Peptide Derived from the Primary Sequence of a Kallikrein Inhibitor Isolated from Bauhinia bauhinioides (pep-BbKI) in an Asthma–COPD Overlap (ACO) Model. International Journal of Molecular Sciences, 24(14), 11261. https://doi.org/10.3390/ijms241411261