
Translating Molecular Biology Discoveries to Develop Targeted Cancer Interception in Barrett’s Esophagus


Abstract
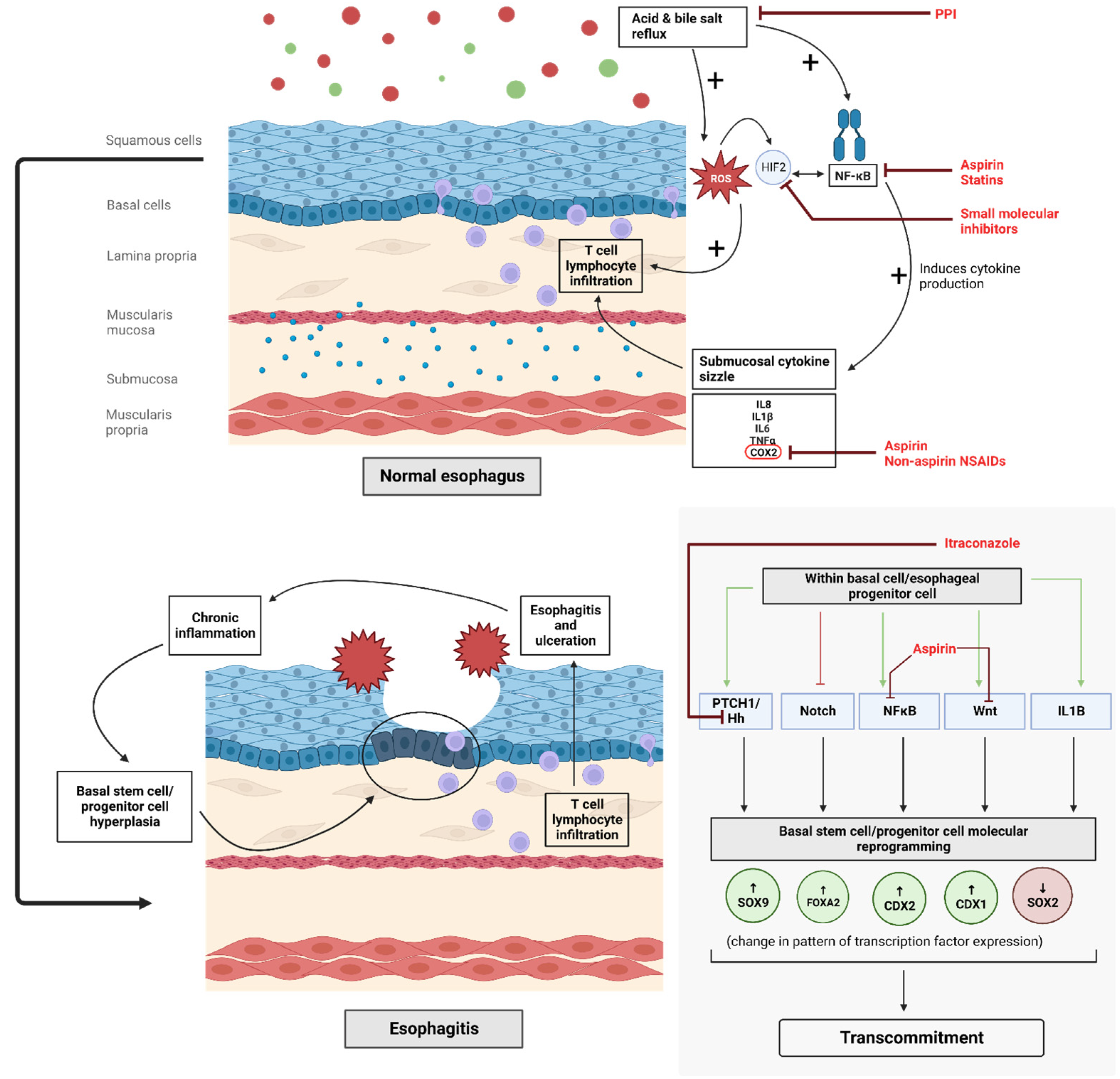
1. Introduction
2. Prevention in Native Barrett’s Esophagus
2.1. Proton Pump Inhibitors
2.2. Non-Steroidal Anti-Inflammatory Drugs and Aspirin
2.3. Metabolic Pathways
2.4. Statins
2.5. Repurposing Cancer Treatment Drugs
2.6. Multipronged Chemoprevention
2.7. Miscellaneous
2.8. Post-Ablation Recurrences of Barrett’s Esophagus

{kind=link}
{kind=link}
| Publication | Type of Study | Study Population | Chemopreventive Agent(s) | Comparison Groups | Endpoints/ Outcomes | Main Results |
|---|---|---|---|---|---|---|
| Abrams et al. (2021) [123] | Randomized, double-blind, placebo-controlled trial | 20 BE patients on continuous PPI therapy | Gastrin/CCK2R antagonist | Netazepide (25 mg/day) vs. placebo | Ki67 density (Ki67 + cells/mm2) as a measure of cellular proliferation Gastrin and CgA levels Gene expression analyses |
|
| Valenzano et al. (2020) [124] | Phase I pilot study | 10 BE patients | Zinc gluconate | Zinc gluconate (52.8 mg/day) vs. placebo | Zinc-induced transcriptional changes in RNA isolated from Barrett’s biopsy |
|
| Jankowski et al. (2018) [24] | Phase III, randomized prospective 2 × 2 factorial trial | 2557 BE patients | PPI, aspirin | High dose esomeprazole (80 mg/day) vs. Low dose esomeprazole (20 mg/day); Aspirin (300/325 mg/day) vs. No aspirin | Composite endpoint of time to all-cause mortality, EA, or HGD (whichever occurs first) |
|
| Cummings et al. (2017) [125] | Multicenter, nonrandomized, interventional pilot study | 18 BE patients | Vitamin D3 | BE cells pre- and post-treatment (Vit D3 50,000 IU/week) vs. normal esophageal cells | Global gene expression, histology, 15-PGDH IHC |
|
| Banerjee et al. (2016) [126] | Open label, single-arm interventional trial | 29 BE patients | UDCA | BE cells pre- and post-treatment (UDCA 13–15 mg/kg/day) | Oxidative DNA damage (8-hydroxydeoxyguanosine levels) Cell proliferation (Ki67 expression) Apoptosis (cleaved caspase 3) |
|
| Bratlie et al. (2016) [127] | Prospective, double-blind, triple-arm, randomized trial | 30 BE patients | ACE inhibitor, AT1R antagonist | High dose PPI (Esomeprazole 40 mg/day) vs. HD PPI + ACEI (Enalapril 5 mg/day) vs. HD PPI + AT1R blocker (Candesartan 8 mg/day) | Expression of proteins known to be associated with inflammation, proliferation, and cancer development (p53, caspase 3, iNOS) |
|
| Chak et al. (2015) [50] | Randomized, double-blind, placebo-controlled trial | 74 BE patients | Metformin | Metformin (increasing to 2000 mg/day) vs. placebo | Change in mean pS6K1 level Cellular proliferation (Ki67 assay) Apoptosis (levels of caspase 3) |
|
| Joe et al. (2015) [128] | Phase 1b Randomized, double-blinded, placebo-controlled dose escalation study | 44 BE patients | Polyphenon E | Poly E (200 mg BID/400 mg BID/600 mg BID) vs. Placebo | Esophageal tissue levels of catechins Endoscopic measurement of BE |
|
| Peng et al. (2014) [129] | 21 BE patients | UDCA | Perfusion of BE cells with DCA and UDCA vs. 24-h pretreatment of BE cells with UDCA followed by perfusion with DCA vs. Pretreatment with oral UDCA (10 mg/kg) followed by perfusion with DCA | Molecular analysis of BE cells |
| |
| Falk et al. (2012) [130] | Phase 2, multicenter, randomized, double-blind, placebo-controlled trial | 122 BE patients | Aspirin | Aspirin placebo + PPI vs. Lower dose aspirin + PPI vs. Higher dose aspirin + PPI | Absolute change in mean tissue PGE2 concentration |
|
| Rawat et al. (2012) [131] | In vitro and in vivo pilot study | 36 BE patients | Curcumin | Curcumin tablet (500 mg/day) Vs. No medication | IL-8 and I-κB gene expression |
|
| de Bortoli et al. (2011) [132] | Single-center, open-label, randomized, parallel group design trial | 77 BE patients | PPI | Esomeprazole (80 mg/day) vs. Pantoprazole (80 mg/day) | Cell proliferation (Ki67 expression) COX-2 expression Apoptosis (TUNEL detection) |
|
| Babar et al. (2010) [133] | Pilot, translational, proof-of-concept trial | 25 BE patients on continuous PPI therapy | Vitamin C | BE cells pre- and post-treatment (Redoxon 1000 mg/day) | NF-kappa B activation Cytokine profile (VEGF, IL8, IL1α, IL1β) |
|
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Islami, F.; DeSantis, C.E.; Jemal, A. Incidence Trends of Esophageal and Gastric Cancer Subtypes by Race, Ethnicity, and Age in the United States, 1997–2014. Clin. Gastroenterol. Hepatol. 2019, 17, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J.; Souza, R.F. Barrett’s Esophagus. N. Engl. J. Med. 2014, 371, 836–845. [Google Scholar] [CrossRef]
- Mukaisho, K.; Kanai, S.; Kushima, R.; Nakayama, T.; Hattori, T.; Sugihara, H. Barretts’s Carcinogenesis. Pathol. Int. 2019, 69, pin.12804. [Google Scholar] [CrossRef]
- Spechler, S.J.; Fitzgerald, R.C.; Prasad, G.A.; Wang, K.K. History, Molecular Mechanisms, and Endoscopic Treatment of Barrett’s Esophagus. Gastroenterology 2010, 138, 854–869. [Google Scholar] [CrossRef]
- Fitzgerald, R.C. Molecular Basis of Barrett’s Oesophagus and Oesophageal Adenocarcinoma. Gut 2006, 55, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Cancer Interception. Cancer Prev. Res. 2011, 4, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F. The Role of Acid and Bile Reflux in Oesophagitis and Barrett’s Metaplasia. Biochem. Soc. Trans. 2010, 38, 348–352. [Google Scholar] [CrossRef]
- Kauer, W.K.H.; Stein, H.J. Role of Acid and Bile in the Genesis of Barrett’s Esophagus. Chest Surg. Clin. N. Am. 2002, 12, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Schlottmann, F.; Molena, D.; Patti, M.G. Gastroesophageal Reflux and Barrett’s Esophagus: A Pathway to Esophageal Adenocarcinoma. Updat. Surg. 2018, 70, 339–342. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Zhou, J.; Liu, P.; Huang, M.; Gunewardena, S.; Mathur, S.C.; Christenson, L.K.; Sharma, M.; Zhang, Q.; Bansal, A. Forkhead Box F1 Induces Columnar Phenotype and Epithelial-to-Mesenchymal Transition in Esophageal Squamous Cells to Initiate Barrett’s like Metaplasia. Lab. Investig. 2021, 101, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Agoston, A.T.; Pham, T.H.; Zhang, W.; Zhang, X.; Huo, X.; Peng, S.; Bajpai, M.; Das, K.; Odze, R.D.; et al. Acidic Bile Salts Induce Epithelial to Mesenchymal Transition via VEGF Signaling in Non-Neoplastic Barrett’s Cells. Gastroenterology 2019, 156, 130–144.e10. [Google Scholar] [CrossRef]
- Souza, R.F.; Huo, X.; Mittal, V.; Schuler, C.M.; Carmack, S.W.; Zhang, H.Y.; Zhang, X.; Yu, C.; Hormi–Carver, K.; Genta, R.M.; et al. Gastroesophageal Reflux Might Cause Esophagitis Through a Cytokine-Mediated Mechanism Rather Than Caustic Acid Injury. Gastroenterology 2009, 137, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, K.B.; Agoston, A.T.; Odze, R.D.; Huo, X.; Pham, T.H.; Cipher, D.J.; Castell, D.O.; Genta, R.M.; Souza, R.F.; Spechler, S.J. Association of Acute Gastroesophageal Reflux Disease with Esophageal Histologic Changes. JAMA 2016, 315, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Agoston, A.T.; Dunbar, K.B.; Cipher, D.J.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Pham, T.H.; Tambar, U.K.; et al. Hypoxia-Inducible Factor-2α Plays a Role in Mediating Oesophagitis in GORD. Gut 2017, 66, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Garg, S.K.; Singh, P.P.; Iyer, P.G.; El-Serag, H.B. Acid-Suppressive Medications and Risk of Oesophageal Adenocarcinoma in Patients with Barrett’s Oesophagus: A Systematic Review and Meta-Analysis. Gut 2014, 63, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthi, R.; Singh, S.; Ragunathan, K.; Visrodia, K.; Wang, K.K.; Katzka, D.A.; Iyer, P.G. Factors Associated with Progression of Barrett’s Esophagus: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 1046–1055.e8. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, C.; Wu, Y.; Chen, X.; Kailas, S.; Karadsheh, Z.; Li, G.; Guo, Z.; Yang, H.; Hu, L.; et al. Do Proton Pump Inhibitors Prevent Barrett’s Esophagus Progression to High-Grade Dysplasia and Esophageal Adenocarcinoma? An Updated Meta-Analysis. J. Cancer Res. Clin. Oncol. 2021, 147, 2681–2691. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, T.-T.; Hong, J.; Fang, J.-Y.; Xiong, H.; Meltzer, S.J. Proton Pump Inhibitors Do Not Reduce the Risk of Esophageal Adenocarcinoma in Patients with Barrett’s Esophagus: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169691. [Google Scholar] [CrossRef]
- Kasiri, K.; Sherwin, C.M.T.; Rostamian, S.; Heidari-Soureshjani, S. Assessment of the Relationship Between Gastric-Acid Suppressants and the Risk of Esophageal Adenocarcinoma: A Systematic Review and Meta-Analysis. Curr. Ther. Res. 2023, 98, 100692. [Google Scholar] [CrossRef]
- Li, L.; Cao, Z.; Zhang, C.; Pan, W. Risk of Esophageal Adenocarcinoma in Patients with Barrett’s Esophagus Using Proton Pump Inhibitors: A Systematic Review with Meta-Analysis and Sequential Trial Analysis. Transl. Cancer Res. 2021, 10, 1620–1627. [Google Scholar] [CrossRef]
- Yao, H.; Wang, L.; Li, H.; Xu, S.; Bai, Z.; Wu, Y.; Chen, H.; Goyal, H.; Qi, X. Proton Pump Inhibitors May Reduce the Risk of High-Grade Dysplasia and/or Esophageal Adenocarcinoma in Barrett’s Esophagus: A Systematic Review and Meta-Analysis. Expert Rev. Clin. Pharmacol. 2022, 15, 79–88. [Google Scholar] [CrossRef]
- Targownik, L.E.; Fisher, D.A.; Saini, S.D. AGA Clinical Practice Update on De-Prescribing of Proton Pump Inhibitors: Expert Review. Gastroenterology 2022, 162, 1334–1342. [Google Scholar] [CrossRef]
- Jankowski, J.A.Z.; De Caestecker, J.; Love, S.B.; Reilly, G.; Watson, P.; Sanders, S.; Ang, Y.; Morris, D.; Bhandari, P.; Brooks, C.; et al. Esomeprazole and Aspirin in Barrett’s Oesophagus (AspECT): A Randomised Factorial Trial. Lancet 2018, 392, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Kandil, H.M.; Tanner, G.; Smalley, W.; Halter, S.; Radhika, A.; Dubois, R.N. Cyclooxygenase-2 Expression in Barrett’s Esophagus. Dig. Dis. Sci. 2001, 46, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.D.; Armstrong, G.R.; Bigley, G.; Green, H.; Attwood, S.E.A. Cyclooxygenase-2 Expression in The Barrettʼs Metaplasia–Dysplasia–Adenocarcinoma Sequence. Am. J. Gastroenterol. 2001, 96, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.T.; Fu, S.; Ramanujam, K.S.; Meltzer, S.J. Increased Expression of Inducible Nitric Oxide Synthase and Cyclooxygenase-2 in Barrett’s Esophagus and Associated Adenocarcinomas. Cancer Res. 1998, 58, 2929–2934. [Google Scholar]
- Zimmermann, K.C.; Sarbia, M.; Weber, A.A.; Borchard, F.; Gabbert, H.E.; Schrör, K. Cyclooxygenase-2 Expression in Human Esophageal Carcinoma. Cancer Res. 1999, 59, 198–204. [Google Scholar]
- Husain, S.S.; Szabo, I.L.; Tarnawski, A.S. NSAID Inhibition of GI Cancer Growth: Clinical Implications and Molecular Mechanisms of Action. Am. J. Gastroenterol. 2002, 97, 542–553. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Q.; Ma, W.-Y.; Wang, K.; Chang, X.; Johnson, M.L.; Bai, R.; Bode, A.M.; Foster, N.R.; Falk, G.W.; et al. Targeting the COX1/2-Driven Thromboxane A2 Pathway Suppresses Barrett’s Esophagus and Esophageal Adenocarcinoma Development. eBioMedicine 2019, 49, 145–156. [Google Scholar] [CrossRef]
- Souza, R.F.; Shewmake, K.; Beer, D.G.; Cryer, B.; Spechler, S.J. Selective Inhibition of Cyclooxygenase-2 Suppresses Growth and Induces Apoptosis in Human Esophageal Adenocarcinoma Cells. Cancer Res. 2000, 60, 5767–5772. [Google Scholar] [PubMed]
- Buttar, N.S. The Effect of Selective Cyclooxygenase-2 Inhibition in Barrett’s Esophagus Epithelium: An In Vitro Study. CancerSpectrum Knowl. Environ. 2002, 94, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Buttar, N.S.; Wang, K.K.; Leontovich, O.; Westcott, J.Y.; Pacifico, R.J.; Anderson, M.A.; Krishnadath, K.K.; Lutzke, L.S.; Burgart, L.J. Chemoprevention of Esophageal Adenocarcinoma by COX-2 Inhibitors in an Animal Model of Barrett’s Esophagus. Gastroenterology 2002, 122, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, P.C.; Oman, K.M.; Paulson, T.G.; Sanchez, C.A.; Zhang, Q.; Marty, J.A.; Delrow, J.J.; Kuhner, M.K.; Vaughan, T.L.; Reid, B.J.; et al. NSAID Use and Somatic Exomic Mutations in Barrett’s Esophagus. Genom. Med. 2018, 10, 17. [Google Scholar] [CrossRef]
- Gammon, M.D.; Terry, M.B.; Arber, N.; Chow, W.-H.; Risch, H.A.; Vaughan, T.L.; Schoenberg, J.B.; Mayne, S.T.; Stanford, J.L.; Dubrow, R.; et al. Nonsteroidal Anti-Inflammatory Drug Use Associated with Reduced Incidence of Adenocarcinomas of the Esophagus and Gastric Cardia That Overexpress Cyclin D1. Cancer Epidemiol. Biomark. Prev. 2004, 13, 34–39. [Google Scholar] [CrossRef]
- Wang, F.; Lv, Z.S.; Fu, Y.K. Nonsteroidal Anti-Inflammatory Drugs and Esophageal Inflammation—Barrett’s Esophagus—Adenocarcinoma Sequence: A Meta-Analysis: Meta-Analysis of Cancer Chemoprevention. Dis. Esophagus 2011, 24, 318–324. [Google Scholar] [CrossRef]
- Liao, L.M.; Vaughan, T.L.; Corley, D.A.; Cook, M.B.; Casson, A.G.; Kamangar, F.; Abnet, C.C.; Risch, H.A.; Giffen, C.; Freedman, N.D.; et al. Nonsteroidal Anti-Inflammatory Drug Use Reduces Risk of Adenocarcinomas of the Esophagus and Esophagogastric Junction in a Pooled Analysis. Gastroenterology 2012, 142, 442–452.e5. [Google Scholar] [CrossRef]
- Schneider, J.L.; Zhao, W.K.; Corley, D.A. Aspirin and Nonsteroidal Anti-Inflammatory Drug Use and the Risk of Barrett’s Esophagus. Dig. Dis. Sci. 2015, 60, 436–443. [Google Scholar] [CrossRef]
- Heath, E.I.; Canto, M.I.; Piantadosi, S.; Montgomery, E.; Weinstein, W.M.; Herman, J.G.; Dannenberg, A.J.; Yang, V.W.; Shar, A.O.; Hawk, E.; et al. Secondary Chemoprevention of Barrett’s Esophagus with Celecoxib: Results of a Randomized Trial. J. Natl. Cancer Inst. 2007, 99, 545–557. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Dunbar, K.B.; Pham, T.H.; Lynch, J.P.; Wang, D.H.; Bresalier, R.S.; et al. Aspirin Prevents NF-ΚB Activation and CDX2 Expression Stimulated by Acid and Bile Salts in Oesophageal Squamous Cells of Patients with Barrett’s Oesophagus. Gut 2017, 67, 606–615. [Google Scholar] [CrossRef]
- Dunbar, K.; Valanciute, A.; Lima, A.C.S.; Vinuela, P.F.; Jamieson, T.; Rajasekaran, V.; Blackmur, J.; Ochocka-Fox, A.-M.; Guazzelli, A.; Cammareri, P.; et al. Aspirin Rescues Wnt-Driven Stem-like Phenotype in Human Intestinal Organoids and Increases the Wnt Antagonist Dickkopf-1. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 465–489. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, Obesity and Cancer: Epidemiological Evidence and Proposed Mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kvapil, P.; Hacken-Bitar, J.; Kramer, J.R. Abdominal Obesity and the Risk of Barrett’s Esophagus. Am. J. Gastroenterol. 2005, 100, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, A.N.; Murad, M.H.; Buttar, N.S.; El–Serag, H.B.; Katzka, D.A.; Iyer, P.G. Central Adiposity Is Associated With Increased Risk of Esophageal Inflammation, Metaplasia, and Adenocarcinoma: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 1399–1412.e7. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Kubo, A.; Levin, T.R.; Block, G.; Habel, L.; Zhao, W.; Leighton, P.; Quesenberry, C.; Rumore, G.J.; Buffler, P.A. Abdominal Obesity and Body Mass Index as Risk Factors for Barrett’s Esophagus. Gastroenterology 2007, 133, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, Z.R.; Farrow, D.C.; Bronner, M.P.; Rosen, S.N.; Vaughan, T.L. Central Adiposity and Risk of Barrett’s Esophagus. Gastroenterology 2007, 133, 403–411. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hashmi, A.; Garcia, J.; Richardson, P.; Alsarraj, A.; Fitzgerald, S.; Vela, M.; Shaib, Y.; Abraham, N.S.; Velez, M.; et al. Visceral Abdominal Obesity Measured by CT Scan Is Associated with an Increased Risk of Barrett’s Oesophagus: A Case-Control Study. Gut 2014, 63, 220–229. [Google Scholar] [CrossRef]
- Cunha Júnior, A.D.; Bragagnoli, A.C.; Costa, F.O.; Carvalheira, J.B.C. Repurposing Metformin for the Treatment of Gastrointestinal Cancer. World J. Gastroenterol. 2021, 27, 1883–1904. [Google Scholar] [CrossRef]
- Chak, A.; Buttar, N.S.; Foster, N.R.; Seisler, D.K.; Marcon, N.E.; Schoen, R.; Cruz-Correa, M.R.; Falk, G.W.; Sharma, P.; Hur, C.; et al. Metformin Does Not Reduce Markers of Cell Proliferation in Esophageal Tissues of Patients with Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2015, 13, 665–672.e4. [Google Scholar] [CrossRef]
- Yendamuri, S.; Barbi, J.; Pabla, S.; Petrucci, C.; Punnanitinont, A.; Nesline, M.; Glenn, S.T.; Depietro, P.; Papanicalou-Sengos, A.; Morrison, C.; et al. Body Mass Index Influences the Salutary Effects of Metformin on Survival After Lobectomy for Stage I NSCLC. J. Thorac. Oncol. 2019, 14, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine Dysregulation and Adipose Tissue Inflammation in Human Obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose Tissue Inflammation and Metabolic Dysfunction in Obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Splenser, A.E.; Kramer, J.; Alsarraj, A.; Fitzgerald, S.; Ramsey, D.; El–Serag, H.B. Circulating Inflammatory Cytokines and Adipokines Are Associated with Increased Risk of Barrett’s Esophagus: A Case–Control Study. Clin. Gastroenterol. Hepatol. 2014, 12, 229–238.e3. [Google Scholar] [CrossRef] [PubMed]
- Kendall, B.J.; Macdonald, G.A.; Hayward, N.K.; Prins, J.B.; Brown, I.; Walker, N.; Pandeya, N.; Green, A.C.; Webb, P.M.; Whiteman, D.C.; et al. Leptin and the Risk of Barrett’s Oesophagus. Gut 2007, 57, 448–454. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Morgenstern, H.; McConell, D.; Scheiman, J.M.; Schoenfeld, P.; Appelman, H.; McMahon, L.F.; Kao, J.Y.; Metko, V.; Zhang, M.; et al. Associations of Diabetes Mellitus, Insulin, Leptin, and Ghrelin With Gastroesophageal Reflux and Barrett’s Esophagus. Gastroenterology 2013, 145, 1237–1244.e5. [Google Scholar] [CrossRef]
- Thompson, O.M.; Beresford, S.A.A.; Kirk, E.A.; Bronner, M.P.; Vaughan, T.L. Serum Leptin and Adiponectin Levels and Risk of Barrett’s Esophagus and Intestinal Metaplasia of the Gastroesophageal Junction. Obesity 2010, 18, 2204–2211. [Google Scholar] [CrossRef]
- Duggan, C.; Onstad, L.; Hardikar, S.; Blount, P.L.; Reid, B.J.; Vaughan, T.L. Association Between Markers of Obesity and Progression From Barrett’s Esophagus to Esophageal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 934–943. [Google Scholar] [CrossRef]
- Mongan, A.M.; Lynam-Lennon, N.; Doyle, S.L.; Casey, R.; Carr, E.; Cannon, A.; Conroy, M.J.; Pidgeon, G.P.; Brennan, L.; Lysaght, J.; et al. Visceral Adipose Tissue Modulates Radiosensitivity in Oesophageal Adenocarcinoma. Int. J. Med. Sci. 2019, 16, 519–528. [Google Scholar] [CrossRef]
- Trevellin, E.; Scarpa, M.; Carraro, A.; Lunardi, F.; Kotsafti, A.; Porzionato, A.; Saadeh, L.; Cagol, M.; Alfieri, R.; Tedeschi, U.; et al. Esophageal Adenocarcinoma and Obesity: Peritumoral Adipose Tissue Plays a Role in Lymph Node Invasion. Oncotarget 2015, 6, 11203–11215. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.H.; Collie-Duguid, E.; Murray, G.I.; Gilbert, F.J.; Denison, A.; Mckiddie, F.; Ahearn, T.; Fleming, I.; Leeds, J.; Phull, P.; et al. Tumour Expression of Leptin Is Associated with Chemotherapy Resistance and Therapy-Independent Prognosis in Gastro-Oesophageal Adenocarcinomas. Br. J. Cancer 2014, 110, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Beales, I.L.P.; Garcia-Morales, C.; Ogunwobi, O.O.; Mutungi, G. Adiponectin Inhibits Leptin-Induced Oncogenic Signalling in Oesophageal Cancer Cells by Activation of PTP1B. Mol. Cell. Endocrinol. 2014, 382, 150–158. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L.P. Leptin Stimulates the Proliferation of Human Oesophageal Adenocarcinoma Cells via HB-EGF and TGFα Mediated Transactivation of the Epidermal Growth Factor Receptor. Br. J. Biomed. Sci. 2008, 65, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Beales, I.L.; Ogunwobi, O.; Cameron, E.; El-Amin, K.; Mutungi, G.; Wilkinson, M. Activation of Akt Is Increased in the Dysplasia-Carcinoma Sequence in Barrett’s Oesophagus and Contributes to Increased Proliferation and Inhibition of Apoptosis: A Histopathological and Functional Study. BMC Cancer 2007, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.; Jain, S. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Fantuzzi, G. Adiponectin in Inflammatory and Immune-Mediated Diseases. Cytokine 2013, 64, 1–10. [Google Scholar] [CrossRef]
- Li, F.; Lam, K.; Xu, A. Therapeutic Perspectives for Adiponectin: An Update. Curr. Med. Chem. 2012, 19, 5513–5523. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L.P. Globular Adiponectin, Acting via Adiponectin Receptor-1, Inhibits Leptin-Stimulated Oesophageal Adenocarcinoma Cell Proliferation. Mol. Cell. Endocrinol. 2008, 285, 43–50. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Dahlkemper, A.; Kao, J.Y.; Zhang, M.; Morgenstern, H.; McMahon, L.; Inadomi, J.M. A Pilot Study of the Association of Low Plasma Adiponectin and Barrett’s Esophagus. Am. J. Gastroenterol. 2008, 103, 1358–1364. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Kao, J.Y.; Madanick, R.D.; Zhang, M.; Wang, M.; Spacek, M.B.; Donovan, J.L.; Bright, S.D.; Shaheen, N.J. Association of Adiponectin Multimers with Barrett’s Oesophagus. Gut 2009, 58, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.-H.; Yang, W.-S.; Liou, J.-M.; Lee, Y.-C.; Wang, H.-P.; Lin, J.-T.; Wu, M.-S. Associations of Circulating Gut Hormone and Adipocytokine Levels with the Spectrum of Gastroesophageal Reflux Disease. PLoS ONE 2015, 10, e0141410. [Google Scholar] [CrossRef] [PubMed]
- Greer, K.B.; Falk, G.W.; Bednarchik, B.; Li, L.; Chak, A. Associations of Serum Adiponectin and Leptin With Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2015, 13, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Almers, L.M.; Graham, J.E.; Havel, P.J.; Corley, D.A. Adiponectin May Modify the Risk of Barrett’s Esophagus in Patients with Gastroesophageal Reflux Disease. Clin. Gastroenterol. Hepatol. 2015, 13, 2256–2264.e3. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The Role of Adiponectin in Cancer: A Review of Current Evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef]
- Alexandre, L. Pathophysiological Mechanisms Linking Obesity and Esophageal Adenocarcinoma. World J. Gastrointest. Pathophysiol. 2014, 5, 534–549. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, J.; Liu, D.; Shan, H.; Zhang, J. Anti-Inflammatory Effect of Full-Length Adiponectin and Proinflammatory Effect of Globular Adiponectin in Esophageal Adenocarcinoma Cells. Oncol. Res. 2013, 21, 15–21. [Google Scholar] [CrossRef]
- Lim, S.; Quon, M.J.; Koh, K.K. Modulation of Adiponectin as a Potential Therapeutic Strategy. Atherosclerosis 2014, 233, 721–728. [Google Scholar] [CrossRef]
- Cho, S.Y.; Park, P.J.; Shin, H.J.; Kim, Y.-K.; Shin, D.W.; Shin, E.S.; Lee, H.H.; Lee, B.G.; Baik, J.-H.; Lee, T.R. (−)-Catechin Suppresses Expression of Kruppel-like Factor 7 and Increases Expression and Secretion of Adiponectin Protein in 3T3-L1 Cells. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1166–E1172. [Google Scholar] [CrossRef]
- Miele, M.; Costantini, S.; Colonna, G. Structural and Functional Similarities between Osmotin from Nicotiana Tabacum Seeds and Human Adiponectin. PLoS ONE 2011, 6, e16690. [Google Scholar] [CrossRef]
- Beales, I.L.P.; Ogunwobi, O.O. Leptin Activates Akt in Oesophageal Cancer Cells via Multiple Atorvastatin-Sensitive Small GTPases. Mol. Cell Biochem. 2021, 476, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L.; Haspinger, E.; La Russa, F.; Maspero, F.; Graziano, P.; Kovalszky, I.; Lovas, S.; Nama, K.; Hoffmann, R.; Knappe, D.; et al. Design and Development of a Peptide-Based Adiponectin Receptor Agonist for Cancer Treatment. BMC Biotechnol. 2011, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L.; Kovalszky, I.; Olah, J.; Coroniti, R.; Knappe, D.; Nollmann, F.I.; Hoffmann, R.; Wade, J.D.; Lovas, S.; Surmacz, E. Optimization of Adiponectin-derived Peptides for Inhibition of Cancer Cell Growth and Signaling. Biopolymers 2015, 104, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Rene Gonzalez, R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M.L. Leptin-Signaling Inhibition Results in Efficient Anti-Tumor Activity in Estrogen Receptor Positive or Negative Breast Cancer. Breast Cancer Res. 2009, 11, R36. [Google Scholar] [CrossRef] [PubMed]
- Demierre, M.-F.; Higgins, P.D.R.; Gruber, S.B.; Hawk, E.; Lippman, S.M. Statins and Cancer Prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef]
- Singh, S.; Singh, A.G.; Singh, P.P.; Murad, M.H.; Iyer, P.G. Statins Are Associated with Reduced Risk of Esophageal Cancer, Particularly in Patients With Barrett’s Esophagus: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 620–629. [Google Scholar] [CrossRef]
- Sadaria, M.R.; Reppert, A.E.; Yu, J.A.; Meng, X.; Fullerton, D.A.; Reece, T.B.; Weyant, M.J. Statin Therapy Attenuates Growth and Malignant Potential of Human Esophageal Adenocarcinoma Cells. J. Thorac. Cardiovasc. Surg. 2011, 142, 1152–1160. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L.P. Statins Inhibit Proliferation and Induce Apoptosis in Barrett’s Esophageal Adenocarcinoma Cells. Am. J. Gastroenterol. 2008, 103, 825–837. [Google Scholar] [CrossRef]
- Samadder, N.J.; Foster, N.; McMurray, R.P.; Burke, C.A.; Stoffel, E.; Kanth, P.; Das, R.; Cruz-Correa, M.; Vilar, E.; Mankaney, G.; et al. Phase II Trial of Weekly Erlotinib Dosing to Reduce Duodenal Polyp Burden Associated with Familial Adenomatous Polyposis. Gut 2023, 72, 256–263. [Google Scholar] [CrossRef]
- Kumble, S.; Omary, M.; Cartwright, C.; Triadafilopoulos, G. Src Activation in Malignant and Premalignant Epithelia of Barrett’s Esophagus. Gastroenterology 1997, 112, 348–356. [Google Scholar] [CrossRef]
- Singh, S.P.; Lipman, J.; Goldman, H.; Ellis, F.H.; Aizenman, L.; Cangi, M.G.; Signoretti, S.; Chiaur, D.S.; Pagano, M.; Loda, M. Loss or Altered Subcellular Localization of P27 in Barrett’s Associated Adenocarcinoma. Cancer Res. 1998, 58, 1730–1735. [Google Scholar] [PubMed]
- Chu, I.; Sun, J.; Arnaout, A.; Kahn, H.; Hanna, W.; Narod, S.; Sun, P.; Tan, C.-K.; Hengst, L.; Slingerland, J. P27 Phosphorylation by Src Regulates Inhibition of Cyclin E-Cdk2. Cell 2007, 128, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Inge, L.J.; Fowler, A.J.; Paquette, K.M.; Richer, A.L.; Tran, N.; Bremner, R.M. Dasatinib, a Small Molecule Inhibitor of the Src Kinase, Reduces the Growth and Activates Apoptosis in Pre-Neoplastic Barrett’s Esophagus Cell Lines: Evidence for a Noninvasive Treatment of High-Grade Dysplasia. J. Thorac. Cardiovasc. Surg. 2013, 145, 531–538. [Google Scholar] [CrossRef] [PubMed]
- NCT00917384. Available online: https://clinicaltrials.gov/ct2/show/NCT00917384 (accessed on 16 June 2023).
- The Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Consortium; Secrier, M.; Li, X.; De Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Iranzo, J.; Martincorena, I.; Koonin, E.V. Cancer-Mutation Network and the Number and Specificity of Driver Mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Dynamic Changes of Driver Genes’ Mutations across Clinical Stages in Nine Cancer Types. Cancer Med. 2016, 5, 1556–1565. [Google Scholar] [CrossRef]
- Choi, S.E.; Perzan, K.E.; Tramontano, A.C.; Kong, C.Y.; Hur, C. Statins and Aspirin for Chemoprevention in Barrett’s Esophagus: Results of a Cost-Effectiveness Analysis. Cancer Prev. Res. 2014, 7, 341–350. [Google Scholar] [CrossRef]
- Shaheen, N.J.; Sharma, P.; Overholt, B.F.; Wolfsen, H.C.; Sampliner, R.E.; Wang, K.K.; Galanko, J.A.; Bronner, M.P.; Goldblum, J.R.; Bennett, A.E.; et al. Radiofrequency Ablation in Barrett’s Esophagus with Dysplasia. N. Engl. J. Med. 2009, 360, 2277–2288. [Google Scholar] [CrossRef]
- Phoa, K.N.; Pouw, R.E.; Van Vilsteren, F.G.I.; Sondermeijer, C.M.T.; Ten Kate, F.J.W.; Visser, M.; Meijer, S.L.; Van Berge Henegouwen, M.I.; Weusten, B.L.A.M.; Schoon, E.J.; et al. Remission of Barrett’s Esophagus with Early Neoplasia 5 Years After Radiofrequency Ablation With Endoscopic Resection: A Netherlands Cohort Study. Gastroenterology 2013, 145, 96–104. [Google Scholar] [CrossRef]
- Orman, E.S.; Li, N.; Shaheen, N.J. Efficacy and Durability of Radiofrequency Ablation for Barrett’s Esophagus: Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 1245–1255. [Google Scholar] [CrossRef]
- Qumseya, B.J.; Wani, S.; Desai, M.; Qumseya, A.; Bain, P.; Sharma, P.; Wolfsen, H. Adverse Events After Radiofrequency Ablation in Patients with Barrett’s Esophagus: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2016, 14, 1086–1095.e6. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthi, R.; Singh, S.; Ragunathan, K.; Katzka, D.A.; Wang, K.K.; Iyer, P.G. Risk of Recurrence of Barrett’s Esophagus after Successful Endoscopic Therapy. Gastrointest. Endosc. 2016, 83, 1090–1106.e3. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, S.; Bulsiewicz, W.J.; Hathorn, K.E.; Komanduri, S.; Muthusamy, V.R.; Rothstein, R.I.; Wolfsen, H.C.; Lightdale, C.J.; Overholt, B.F.; Camara, D.S.; et al. Durability and Predictors of Successful Radiofrequency Ablation for Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2014, 12, 1840–1847.e1. [Google Scholar] [CrossRef] [PubMed]
- Fujii-Lau, L.; Cinnor, B.; Shaheen, N.; Gaddam, S.; Komanduri, S.; Muthusamy, V.; Das, A.; Wilson, R.; Simon, V.; Kushnir, V.; et al. Recurrence of Intestinal Metaplasia and Early Neoplasia after Endoscopic Eradication Therapy for Barrett’s Esophagus: A Systematic Review and Meta-Analysis. Endosc. Int. Open. 2017, 05, E430–E449. [Google Scholar] [CrossRef]
- Sami, S.S.; Ravindran, A.; Kahn, A.; Snyder, D.; Santiago, J.; Ortiz-Fernandez-Sordo, J.; Tan, W.K.; Dierkhising, R.A.; Crook, J.E.; Heckman, M.G.; et al. Timeline and Location of Recurrence Following Successful Ablation in Barrett’s Oesophagus: An International Multicentre Study. Gut 2019, 68, 1379–1385. [Google Scholar] [CrossRef]
- Cotton, C.C.; Wolf, W.A.; Overholt, B.F.; Li, N.; Lightdale, C.J.; Wolfsen, H.C.; Pasricha, S.; Wang, K.K.; Shaheen, N.J.; Sampliner, R.E.; et al. Late Recurrence of Barrett’s Esophagus After Complete Eradication of Intestinal Metaplasia Is Rare: Final Report from Ablation in Intestinal Metaplasia Containing Dysplasia Trial. Gastroenterology 2017, 153, 681–688.e2. [Google Scholar] [CrossRef]
- Wang, D.H.; Souza, R.F. Transcommitment: Paving the Way to Barrett’s Metaplasia. In Stem Cells, Pre-Neoplasia, and Early Cancer of the Upper Gastrointestinal Tract; Jansen, M., Wright, N.A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 908, pp. 183–212. ISBN 978-3-319-41386-0. [Google Scholar]
- Wang, D.H.; Clemons, N.J.; Miyashita, T.; Dupuy, A.J.; Zhang, W.; Szczepny, A.; Corcoran–Schwartz, I.M.; Wilburn, D.L.; Montgomery, E.A.; Wang, J.S.; et al. Aberrant Epithelial–Mesenchymal Hedgehog Signaling Characterizes Barrett’s Metaplasia. Gastroenterology 2010, 138, 1810–1822.e2. [Google Scholar] [CrossRef]
- Komanduri, S.; Kahrilas, P.J.; Krishnan, K.; McGorisk, T.; Bidari, K.; Grande, D.; Keefer, L.; Pandolfino, J. Recurrence of Barrett’s Esophagus Is Rare Following Endoscopic Eradication Therapy Coupled with Effective Reflux Control. Am. J. Gastroenterol. 2017, 112, 556–566. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal That Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-Label, Exploratory Phase II Trial of Oral Itraconazole for the Treatment of Basal Cell Carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing Itraconazole as a Treatment for Advanced Prostate Cancer: A Noncomparative Randomized Phase II Trial in Men with Metastatic Castration-Resistant Prostate Cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.E.; Putnam, W.C.; Fattah, F.J.; Kernstine, K.H.; Brekken, R.A.; Pedrosa, I.; Skelton, R.; Saltarski, J.M.; Lenkinski, R.E.; Leff, R.D.; et al. Concentration-Dependent Early Antivascular and Antitumor Effects of Itraconazole in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 6017–6027. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Ansari, A.M.; Miyashita, T.; Zahurak, M.; Lay, F.; Ahmed, A.K.; Born, L.J.; Pezhouh, M.K.; Salimian, K.J.; Ng, C.; et al. Targeting the Hedgehog Pathway Using Itraconazole to Prevent Progression of Barrett’s Esophagus to Invasive Esophageal Adenocarcinoma. Ann. Surg. 2021, 273, e206–e213. [Google Scholar] [CrossRef]
- Chen, M.-B.; Liu, Y.-Y.; Xing, Z.-Y.; Zhang, Z.-Q.; Jiang, Q.; Lu, P.-H.; Cao, C. Itraconazole-Induced Inhibition on Human Esophageal Cancer Cell Growth Requires AMPK Activation. Mol. Cancer Ther. 2018, 17, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bhagwath, A.S.; Ramzan, Z.; Williams, T.A.; Subramaniyan, I.; Edpuganti, V.; Kallem, R.R.; Dunbar, K.B.; Ding, P.; Gong, K.; et al. Itraconazole Exerts Its Antitumor Effect in Esophageal Cancer By Suppressing the HER2/AKT Signaling Pathway. Mol. Cancer Ther. 2021, 20, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- NCT05609253. Available online: https://clinicaltrials.gov/ct2/show/NCT05609253 (accessed on 26 April 2023).
- Zhou, C.; Tsai, T.-H.; Lee, H.-C.; Kirtane, T.; Figueiredo, M.; Tao, Y.K.; Ahsen, O.O.; Adler, D.C.; Schmitt, J.M.; Huang, Q.; et al. Characterization of Buried Glands before and after Radiofrequency Ablation by Using 3-Dimensional Optical Coherence Tomography (with Videos). Gastrointest. Endosc. 2012, 76, 32–40. [Google Scholar] [CrossRef]
- Anders, M.; Lucks, Y.; El-Masry, M.A.; Quaas, A.; Rösch, T.; Schachschal, G.; Bähr, C.; Gauger, U.; Sauter, G.; Izbicki, J.R.; et al. Subsquamous Extension of Intestinal Metaplasia Is Detected in 98% of Cases of Neoplastic Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2014, 12, 405–410. [Google Scholar] [CrossRef]
- Konda, V.; Souza, R.F.; Dunbar, K.B.; Mills, J.C.; Kim, D.S.; Odze, R.D.; Spechler, S.J. An Endoscopic and Histologic Study on Healing of Radiofrequency Ablation Wounds in Patients with Barrett’s Esophagus. Am. J. Gastroenterol. 2022, 117, 1583–1592. [Google Scholar] [CrossRef]
- Zhang, Q.; Bansal, A.; Dunbar, K.B.; Chang, Y.; Zhang, J.; Balaji, U.; Gu, J.; Zhang, X.; Podgaetz, E.; Pan, Z.; et al. A Human Barrett’s Esophagus Organoid System Reveals Epithelial-Mesenchymal Plasticity Induced by Acid and Bile Salts. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G598–G614. [Google Scholar] [CrossRef]
- Abrams, J.A.; Del Portillo, A.; Hills, C.; Compres, G.; Friedman, R.A.; Cheng, B.; Poneros, J.; Lightdale, C.J.; De La Rue, R.; Di Pietro, M.; et al. Randomized Controlled Trial of the Gastrin/CCK2 Receptor Antagonist Netazepide in Patients with Barrett’s Esophagus. Cancer Prev. Res. 2021, 14, 675–682. [Google Scholar] [CrossRef]
- Valenzano, M.C.; Rybakovsky, E.; Chen, V.; Leroy, K.; Lander, J.; Richardson, E.; Yalamanchili, S.; McShane, S.; Mathew, A.; Mayilvaganan, B.; et al. Zinc Gluconate Induces Potentially Cancer Chemopreventive Activity in Barrett’s Esophagus: A Phase 1 Pilot Study. Dig. Dis. Sci. 2021, 66, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Cummings, L.C.; Thota, P.N.; Willis, J.E.; Chen, Y.; Cooper, G.S.; Furey, N.; Bednarchik, B.; Alashkar, B.M.; Dumot, J.; Faulx, A.L.; et al. A Nonrandomized Trial of Vitamin D Supplementation for Barrett’s Esophagus. PLoS ONE 2017, 12, e0184928. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.; Shaheen, N.J.; Martinez, J.A.; Hsu, C.-H.; Trowers, E.; Gibson, B.A.; Della’Zanna, G.; Richmond, E.; Chow, H.-H.S. Clinical Study of Ursodeoxycholic Acid in Barrett’s Esophagus Patients. Cancer Prev. Res. 2016, 9, 528–533. [Google Scholar] [CrossRef]
- Bratlie, S.O.; Casselbrant, A.; Edebo, A.; Fändriks, L. Support for Involvement of the Renin–Angiotensin System in Dysplastic Barrett’s Esophagus. Scand. J. Gastroenterol. 2017, 52, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Joe, A.K.; Schnoll-Sussman, F.; Bresalier, R.S.; Abrams, J.A.; Hibshoosh, H.; Cheung, K.; Friedman, R.A.; Yang, C.S.; Milne, G.L.; Liu, D.D.; et al. Phase Ib Randomized, Double-Blinded, Placebo-Controlled, Dose Escalation Study of Polyphenon E in Patients with Barrett’s Esophagus. Cancer Prev. Res. 2015, 8, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Huo, X.; Rezaei, D.; Zhang, Q.; Zhang, X.; Yu, C.; Asanuma, K.; Cheng, E.; Pham, T.H.; Wang, D.H.; et al. In Barrett’s Esophagus Patients and Barrett’s Cell Lines, Ursodeoxycholic Acid Increases Antioxidant Expression and Prevents DNA Damage by Bile Acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G129–G139. [Google Scholar] [CrossRef] [PubMed]
- Falk, G.W.; Buttar, N.S.; Foster, N.R.; Ziegler, K.L.A.; DeMars, C.J.; Romero, Y.; Marcon, N.E.; Schnell, T.; Corley, D.A.; Sharma, P.; et al. A Combination of Esomeprazole and Aspirin Reduces Tissue Concentrations of Prostaglandin E2 in Patients With Barrett’s Esophagus. Gastroenterology 2012, 143, 917–926.e1. [Google Scholar] [CrossRef] [PubMed]
- Rawat, N.; Alhamdani, A.; McAdam, E.; Cronin, J.; Eltahir, Z.; Lewis, P.; Griffiths, P.; Baxter, J.N.; Jenkins, G.J.S. Curcumin Abrogates Bile-Induced NF-ΚB Activity and DNA Damage in Vitro and Suppresses NF-ΚB Activity Whilst Promoting Apoptosis in Vivo, Suggesting Chemopreventative Potential in Barrett’s Oesophagus. Clin. Transl. Oncol. 2012, 14, 302–311. [Google Scholar] [CrossRef]
- De Bortoli, N.; Martinucci, I.; Piaggi, P.; Maltinti, S.; Bianchi, G.; Ciancia, E.; Gambaccini, D.; Lenzi, F.; Costa, F.; Leonardi, G.; et al. Randomised Clinical Trial: Twice Daily Esomeprazole 40 Mg vs. Pantoprazole 40 Mg in Barrett’s Oesophagus for 1 Year: Randomised Clinical Trial: Double-Dose PPI in Patients with Barrett’s Oesophagus. Aliment. Pharmacol. Ther. 2011, 33, 1019–1027. [Google Scholar] [CrossRef]
- Babar, M.; Abdel-Latif, M.M.M.; Ravi, N.; Murphy, A.; Byrne, P.J.; Kelleher, D.; Reynolds, J.V. Pilot Translational Study of Dietary Vitamin C Supplementation in Barrett’s Esophagus. Dis. Esophagus 2010, 23, 271–276. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samaddar, S.; Buckles, D.; Saha, S.; Zhang, Q.; Bansal, A. Translating Molecular Biology Discoveries to Develop Targeted Cancer Interception in Barrett’s Esophagus. Int. J. Mol. Sci. 2023, 24, 11318. https://doi.org/10.3390/ijms241411318
Samaddar S, Buckles D, Saha S, Zhang Q, Bansal A. Translating Molecular Biology Discoveries to Develop Targeted Cancer Interception in Barrett’s Esophagus. International Journal of Molecular Sciences. 2023; 24(14):11318. https://doi.org/10.3390/ijms241411318
Chicago/Turabian StyleSamaddar, Sohini, Daniel Buckles, Souvik Saha, Qiuyang Zhang, and Ajay Bansal. 2023. "Translating Molecular Biology Discoveries to Develop Targeted Cancer Interception in Barrett’s Esophagus" International Journal of Molecular Sciences 24, no. 14: 11318. https://doi.org/10.3390/ijms241411318
APA StyleSamaddar, S., Buckles, D., Saha, S., Zhang, Q., & Bansal, A. (2023). Translating Molecular Biology Discoveries to Develop Targeted Cancer Interception in Barrett’s Esophagus. International Journal of Molecular Sciences, 24(14), 11318. https://doi.org/10.3390/ijms241411318