
miR-29a-3p Regulates Autophagy by Targeting Akt3-Mediated mTOR in SiO2-Induced Lung Fibrosis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
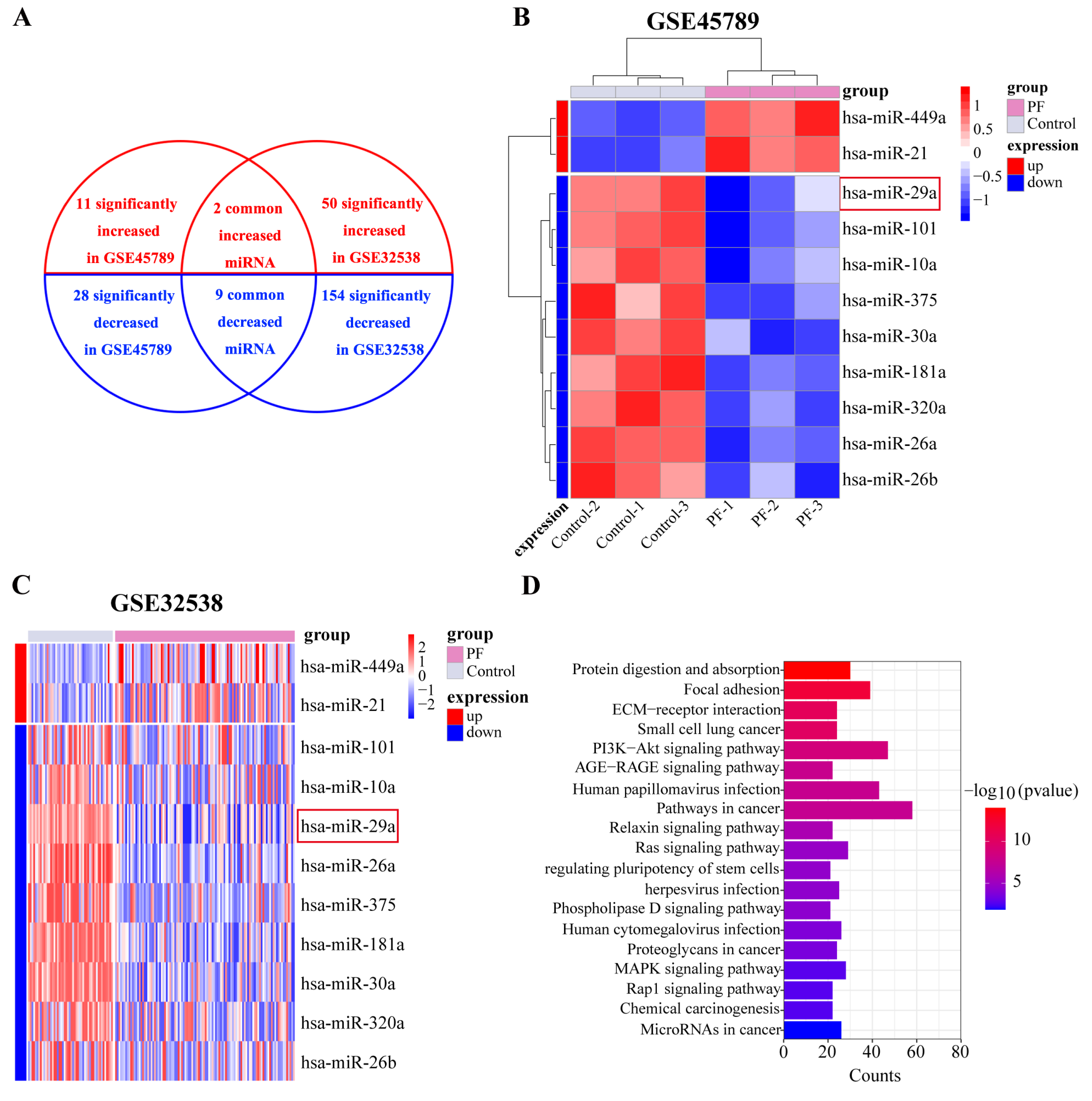
2.1. miR-29a Expression Is Significantly Downregulated in Fibrotic Lung Tissue Based on Bioinformatics Analysis
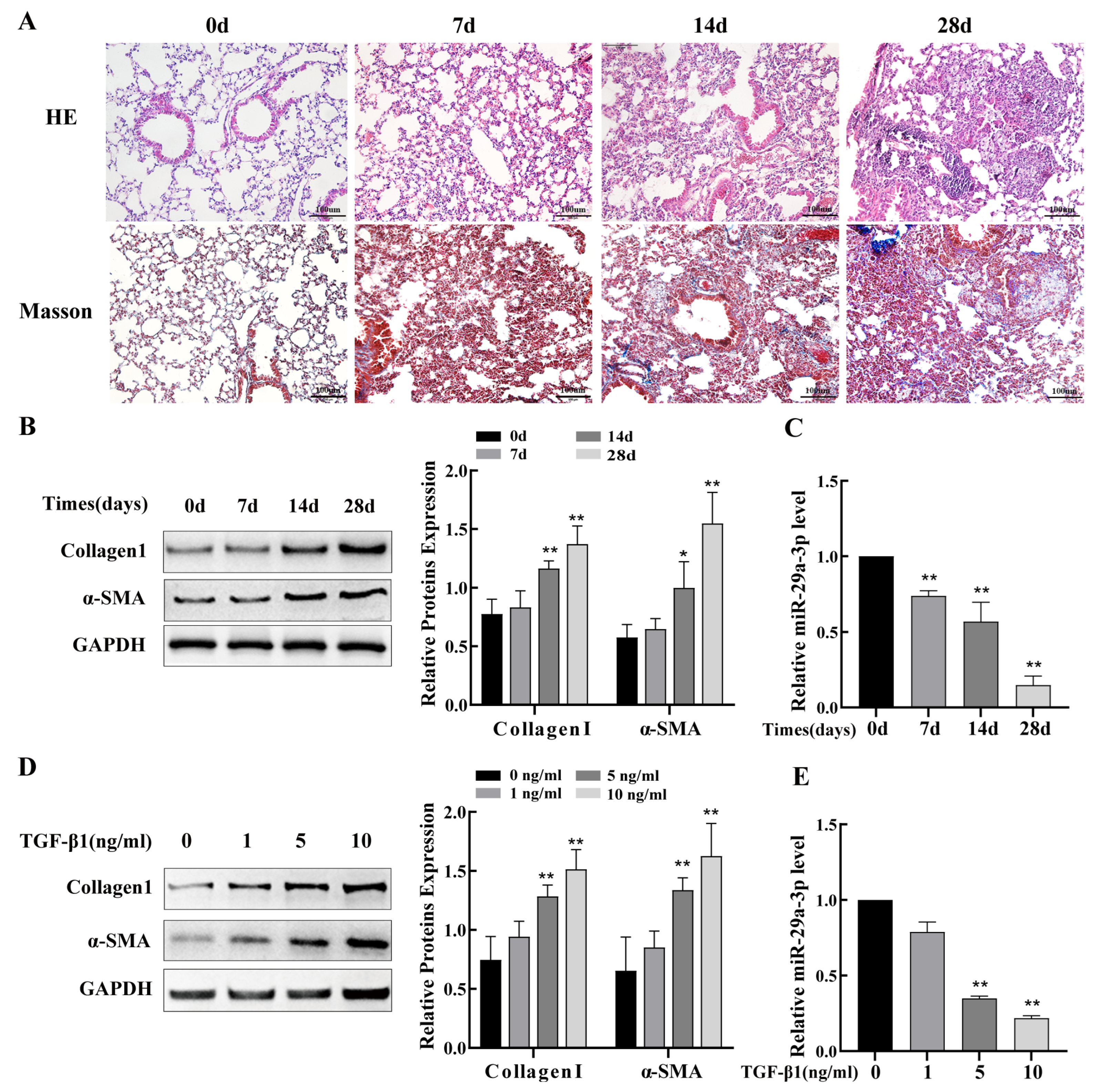
2.2. miR-29a-3p Expression Was Downregulated In Silica-Induced PF Model
2.3. Autophagy Regulates PF Caused by Silica
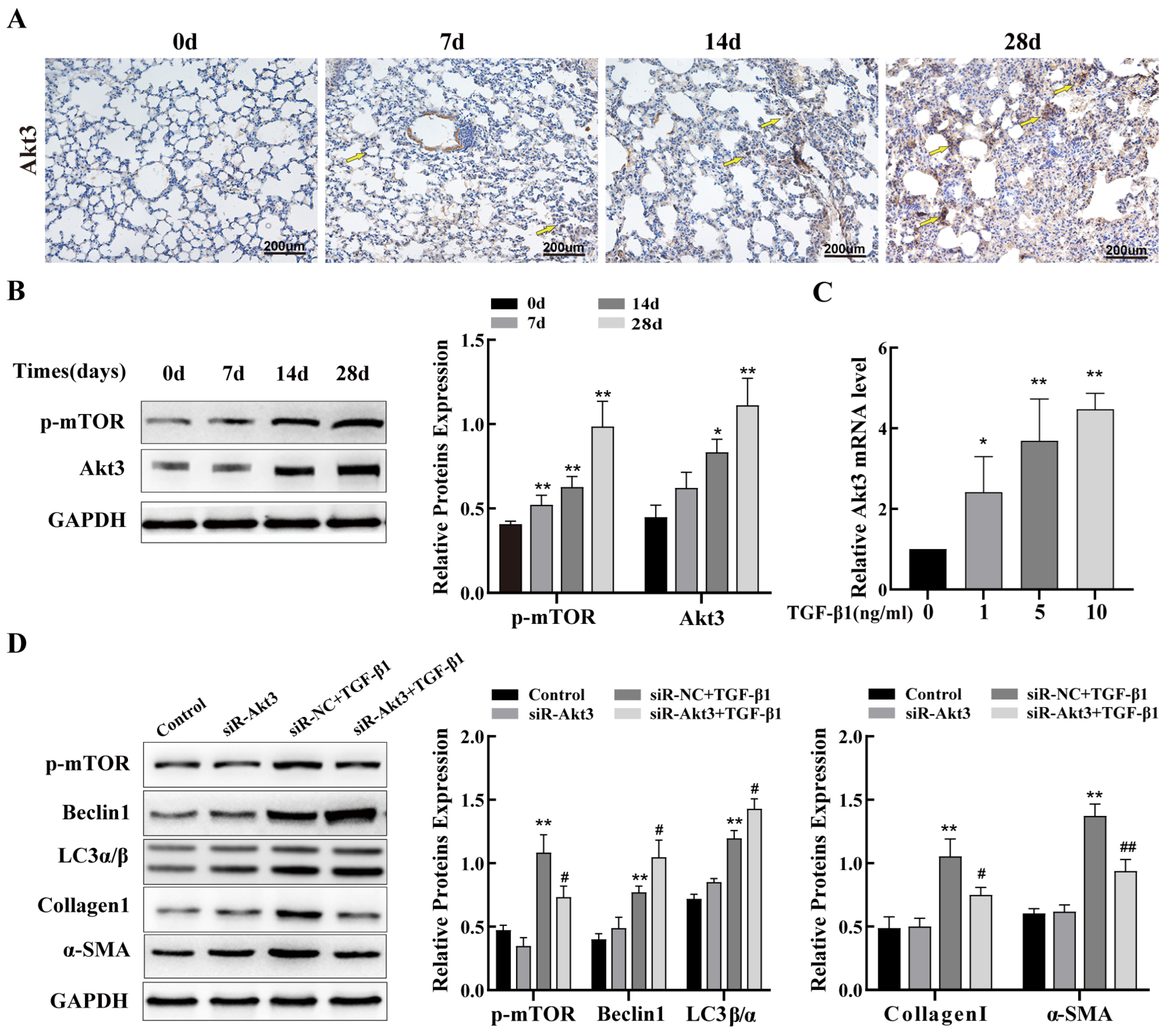
2.4. Inhibition of Akt3 Expression Promotes Autophagy in Lung Epithelial Cells and Regulates Fibrosis
2.5. miR-29a-3p Targets Akt3 and Inhibits Its Expression
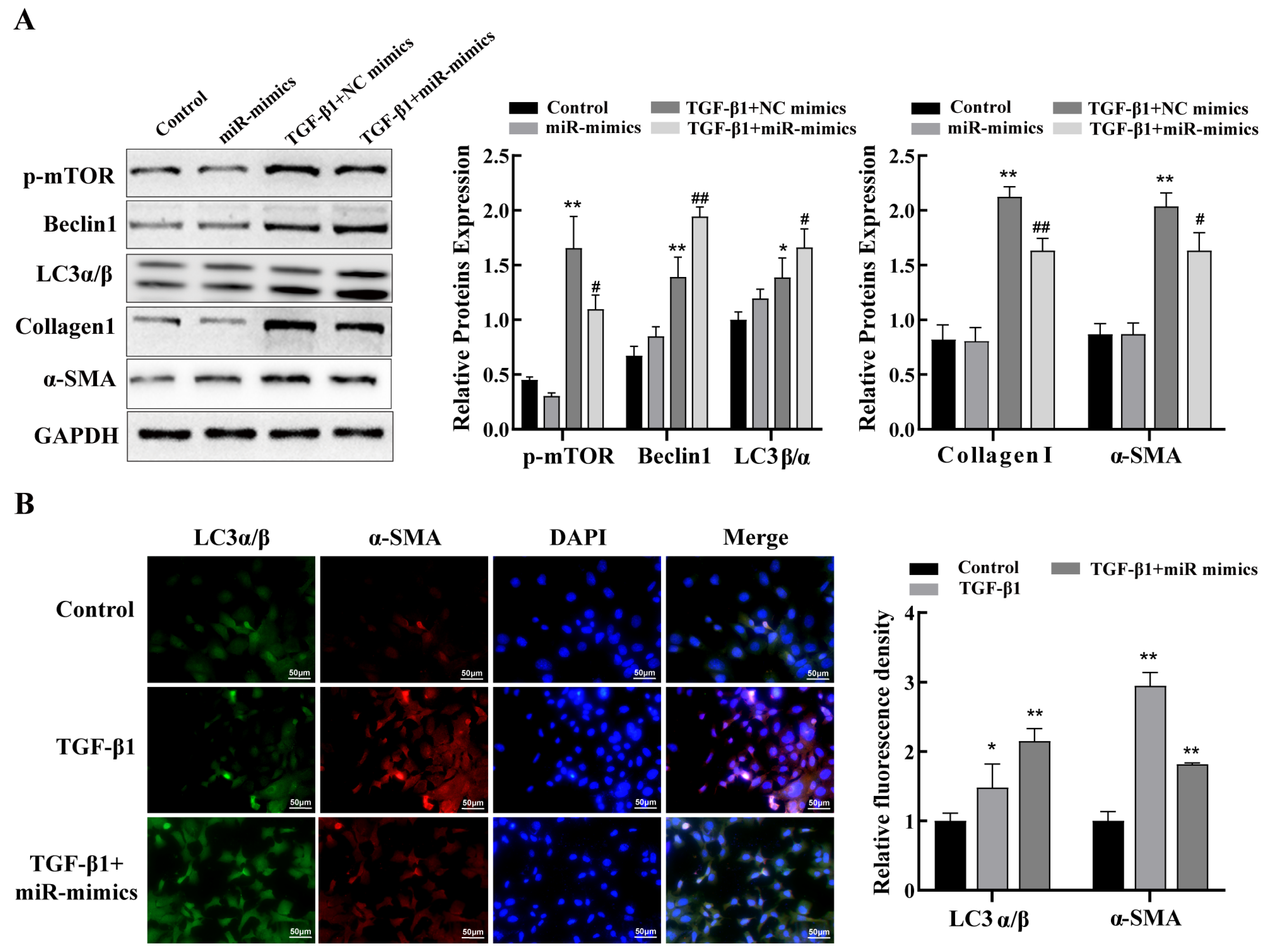
2.6. miR-29a-3p Regulates Autophagy via the Akt3/mTOR Axis to Attenuate Fibrosis In Vitro
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis
4.2. Animals
4.3. Silica-Induced PF Mouse Model
4.4. Histopathological and Immunohistochemistry (IHC) Analysis
4.5. Cell Culture, Plasmid Construction, and Transfection
4.6. RNA Extraction and Quantification
4.7. Western Blotting
4.8. Immunofluorescence
4.9. Detection of Autophagosomes Using Transmission Electron Microscopy (TEM)
4.10. Dual-Luciferase Reporter Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Désogère, P.; Tapias, L.F.; Hariri, L.P.; Rotile, N.J.; Rietz, T.A.; Probst, C.K.; Blasi, F.; Day, H.; Mino-Kenudson, M.; Weinreb, P.; et al. Type I collagen-targeted PET probe for pulmonary fibrosis detection and staging in preclinical models. Sci. Transl. Med. 2017, 9, eaaf4696. [Google Scholar] [CrossRef] [Green Version]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Leung, C.C.; Yu, I.T.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Kramer, M.R.; Blanc, P.D.; Fireman, E.; Amital, A.; Guber, A.; Rhahman, N.A.; Shitrit, D. Artificial stone silicosis [corrected]: Disease resurgence among artificial stone workers. Chest 2012, 142, 419–424. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, M.; Liu, Y.; Ma, J.; Yang, M.; Shi, T.; Chen, W. Comparison of Risk of Silicosis in Metal Mines and Pottery Factories: A 44-Year Cohort Study. Chest 2020, 158, 1050–1059. [Google Scholar] [CrossRef]
- Hoy, R.F.; Chambers, D.C. Silica-related diseases in the modern world. Allergy 2020, 75, 2805–2817. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Qi, X.; Luo, Y.; Li, X.; Shu, T.; Li, B.; Song, M.; Liu, Y.; Wei, D.; Chen, J.; et al. Multi-omics study of silicosis reveals the potential therapeutic targets PGD(2) and TXA(2). Theranostics 2021, 11, 2381–2394. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Qiu, T.; Liu, W.; Yao, P. Autophagy, an important therapeutic target for pulmonary fibrosis diseases. Clin. Chim. Acta 2020, 502, 139–147. [Google Scholar] [CrossRef]
- Du, S.; Li, C.; Lu, Y.; Lei, X.; Zhang, Y.; Li, S.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Alleviates Crystalline Silica-Induced Pulmonary Inflammation and Fibrosis through Promoting Alveolar Macrophage Autophagy. Theranostics 2019, 9, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef]
- Li, N.; Shi, F.; Wang, X.; Yang, P.; Sun, K.; Zhang, L.; Hao, X.; Li, X.; Li, J.; Jin, Y. Silica dust exposure induces pulmonary fibrosis through autophagy signaling. Environ. Toxicol. 2021, 36, 1269–1277. [Google Scholar] [CrossRef]
- Wei, Y.; Kim, T.J.; Peng, D.H.; Duan, D.; Gibbons, D.L.; Yamauchi, M.; Jackson, J.R.; Le Saux, C.J.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J. Clin. Investig. 2017, 127, 3675–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Liu, S.; Chua, M.S.; Li, H.; Luo, D.; Wang, S.; Zhang, S.; Han, B.; Sun, C. SOCS5 inhibition induces autophagy to impair metastasis in hepatocellular carcinoma cells via the PI3K/Akt/mTOR pathway. Cell Death Dis. 2019, 10, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Zhang, T.; Shi, Z.; Wang, Y.; Wang, L.; Zhang, B.; Chen, G.; Wan, Q.; Chen, L. Akt3 deletion in mice impairs spatial cognition and hippocampal CA1 long long-term potentiation through downregulation of mTOR. Acta Physiol. 2019, 225, e13167. [Google Scholar] [CrossRef]
- DuBois, J.C.; Ray, A.K.; Gruber, R.C.; Zhang, Y.; Aflakpui, R.; Macian-Juan, F.; Shafit-Zagardo, B. Akt3-Mediated Protection Against Inflammatory Demyelinating Disease. Front. Immunol. 2019, 10, 1738. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Biswas, S.; Morton, R.E.; Smith, J.D.; Hay, N.; Byzova, T.V.; Febbraio, M.; Podrez, E.A. Akt3 deficiency in macrophages promotes foam cell formation and atherosclerosis in mice. Cell Metab. 2012, 15, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Arranz, A.; Doxaki, C.; Vergadi, E.; Martinez de la Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef]
- Chen, X.; Ariss, M.M.; Ramakrishnan, G.; Nogueira, V.; Blaha, C.; Putzbach, W.; Islam, A.; Frolov, M.V.; Hay, N. Cell-Autonomous versus Systemic Akt Isoform Deletions Uncovered New Roles for Akt1 and Akt2 in Breast Cancer. Mol. Cell 2020, 80, 87–101.e5. [Google Scholar] [CrossRef]
- Li, P.; Yao, Y.; Ma, Y.; Chen, Y. MiR-150 attenuates LPS-induced acute lung injury via targeting AKT3. Int. Immunopharmacol. 2019, 75, 105794. [Google Scholar] [CrossRef]
- Fu, J.; Peng, L.; Tao, T.; Chen, Y.; Li, Z.; Li, J. Regulatory roles of the miR-200 family in neurodegenerative diseases. Biomed. Pharmacother. 2019, 119, 109409. [Google Scholar] [CrossRef] [PubMed]
- Pattarayan, D.; Thimmulappa, R.K.; Ravikumar, V.; Rajasekaran, S. Diagnostic Potential of Extracellular MicroRNA in Respiratory Diseases. Clin. Rev. Allergy Immunol. 2018, 54, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.W.Q.; Sim, W.L.; Cheong, J.K.; Kuan, W.S.; Tran, T.; Lim, H.F. MicroRNAs in chronic airway diseases: Clinical correlation and translational applications. Pharmacol. Res. 2020, 160, 105045. [Google Scholar] [CrossRef] [PubMed]
- Cushing, M.C.; Mariner, P.D.; Liao, J.T.; Sims, E.A.; Anseth, K.S. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. Faseb J. 2008, 22, 1769–1777. [Google Scholar] [CrossRef] [Green Version]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]
- Cong, L.H.; Li, T.; Wang, H.; Wu, Y.N.; Wang, S.P.; Zhao, Y.Y.; Zhang, G.Q.; Duan, J. IL-17A-producing T cells exacerbate fine particulate matter-induced lung inflammation and fibrosis by inhibiting PI3K/Akt/mTOR-mediated autophagy. J. Cell Mol. Med. 2020, 24, 8532–8544. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Yao, W.; Yang, P.; Qi, Y.; Jin, L.; Zhao, A.; Ding, M.; Wang, D.; Li, Y.; Hao, C. Transcriptome analysis reveals a protective role of liver X receptor alpha against silica particle-induced experimental silicosis. Sci. Total Environ. 2020, 747, 141531. [Google Scholar] [CrossRef]
- Gulumian, M.; Borm, P.J.; Vallyathan, V.; Castranova, V.; Donaldson, K.; Nelson, G.; Murray, J. Mechanistically identified suitable biomarkers of exposure, effect, and susceptibility for silicosis and coal-worker’s pneumoconiosis: A comprehensive review. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 357–395. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Li, P.; Pan, H.; Xu, Q.; Xu, T.; Li, Y.; Wei, D.; Mo, Y.; Zhang, Q.; Chen, J.; et al. miR-770-5p inhibits the activation of pulmonary fibroblasts and silica-induced pulmonary fibrosis through targeting TGFBR1. Ecotoxicol. Environ. Saf. 2021, 220, 112372. [Google Scholar] [PubMed]
- Wang, Z.; Wang, C.; Liu, S.; He, W.; Wang, L.; Gan, J.; Huang, Z.; Wang, Z.; Wei, H.; Zhang, J.; et al. Specifically Formed Corona on Silica Nanoparticles Enhances Transforming Growth Factor β1 Activity in Triggering Lung Fibrosis. ACS Nano 2017, 11, 1659–1672. [Google Scholar] [CrossRef]
- Bahudhanapati, H.; Tan, J.; Dutta, J.A.; Strock, S.B.; Sembrat, J.; Àlvarez, D.; Rojas, M.; Jäger, B.; Prasse, A.; Zhang, Y.; et al. MicroRNA-144-3p targets relaxin/insulin-like family peptide receptor 1 (RXFP1) expression in lung fibroblasts from patients with idiopathic pulmonary fibrosis. J. Biol. Chem. 2019, 294, 5008–5022. [Google Scholar] [CrossRef] [Green Version]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Chioccioli, M.; Roy, S.; Newell, R.; Pestano, L.; Dickinson, B.; Rigby, K.; Herazo-Maya, J.; Jenkins, G.; Ian, S.; Saini, G.; et al. A lung targeted miR-29 mimic as a therapy for pulmonary fibrosis. EBioMedicine 2022, 85, 104304. [Google Scholar] [CrossRef]
- Xiao, J.; Meng, X.M.; Huang, X.R.; Chung, A.C.; Feng, Y.L.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol. Ther. 2012, 20, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Baek, A.R.; Lee, J.H.; Jang, A.S.; Kim, D.J.; Chin, S.S.; Park, S.W. IL-37 Attenuates Lung Fibrosis by Inducing Autophagy and Regulating TGF-β1 Production in Mice. J. Immunol. 2019, 203, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yuan, J.; Yao, S.; Jin, Y.; Chen, G.; Tian, W.; Xi, J.; Xu, Z.; Weng, D.; Chen, J. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy 2015, 11, 2346–2357. [Google Scholar] [CrossRef] [Green Version]
- Polytarchou, C.; Hatziapostolou, M.; Yau, T.O.; Christodoulou, N.; Hinds, P.W.; Kottakis, F.; Sanidas, I.; Tsichlis, P.N. Akt3 induces oxidative stress and DNA damage by activating the NADPH oxidase via phosphorylation of p47(phox). Proc. Natl. Acad. Sci. USA 2020, 117, 28806–28815. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Bensinger, S.J. (Sterol)ized Immunity: Could PI3K/AKT3 Be the Answer? Immunity 2020, 52, 4–6. [Google Scholar] [CrossRef]
- Corum, D.G.; Tsichlis, P.N.; Muise-Helmericks, R.C. AKT3 controls mitochondrial biogenesis and autophagy via regulation of the major nuclear export protein CRM-1. Faseb J. 2014, 28, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Cai, B.; Li, X.; Li, D.; Yin, G. MiR-125b-5p and miR-181b-5p inhibit keratinocyte proliferation in skin by targeting Akt3. Eur. J. Pharmacol. 2019, 862, 172659. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Hao, X.; Liu, J.; Zhang, Q.; Liang, Z.; Li, X.; Liu, H. miR-29a-3p Regulates Autophagy by Targeting Akt3-Mediated mTOR in SiO2-Induced Lung Fibrosis. Int. J. Mol. Sci. 2023, 24, 11440. https://doi.org/10.3390/ijms241411440
Li P, Hao X, Liu J, Zhang Q, Liang Z, Li X, Liu H. miR-29a-3p Regulates Autophagy by Targeting Akt3-Mediated mTOR in SiO2-Induced Lung Fibrosis. International Journal of Molecular Sciences. 2023; 24(14):11440. https://doi.org/10.3390/ijms241411440
Chicago/Turabian StyleLi, Peiyuan, Xiaohui Hao, Jiaxin Liu, Qinxin Zhang, Zixuan Liang, Xinran Li, and Heliang Liu. 2023. "miR-29a-3p Regulates Autophagy by Targeting Akt3-Mediated mTOR in SiO2-Induced Lung Fibrosis" International Journal of Molecular Sciences 24, no. 14: 11440. https://doi.org/10.3390/ijms241411440