
Hydration and Structural Adaptations of the Human CYP1A1, CYP1A2, and CYP1B1 Active Sites by Molecular Dynamics Simulations



Abstract
:1. Introduction
2. Results and Discussion
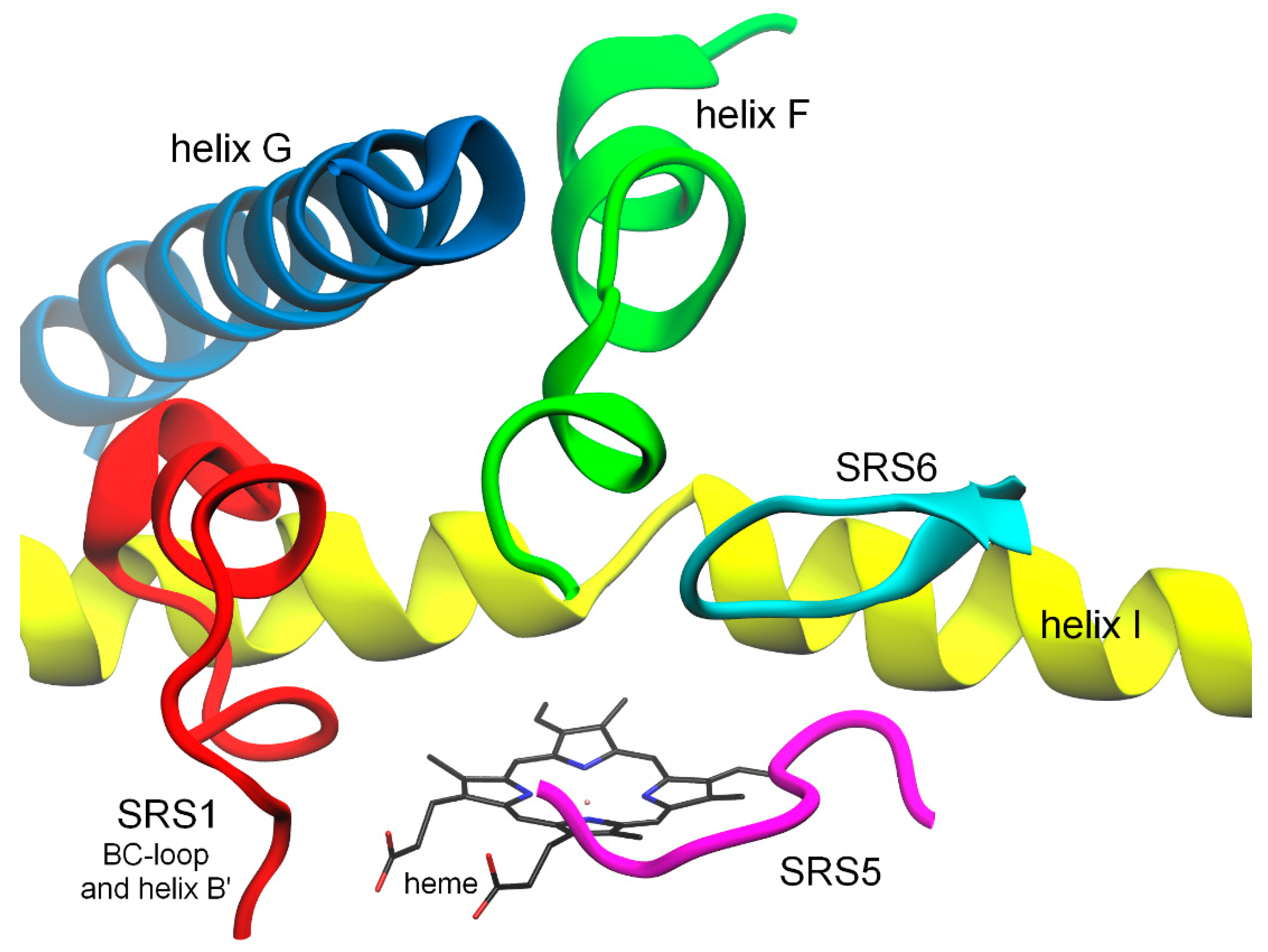
2.1. Structural Changes in Substrate Recognition Sites
2.2. Hydration of CYP1 Structures in APO Forms and as Complexes with the Polymethoxy-trans-Stilbenes
2.2.1. Hydration of the Crystal Structures of CYP1A1, CYP1A2, and CYP1B1
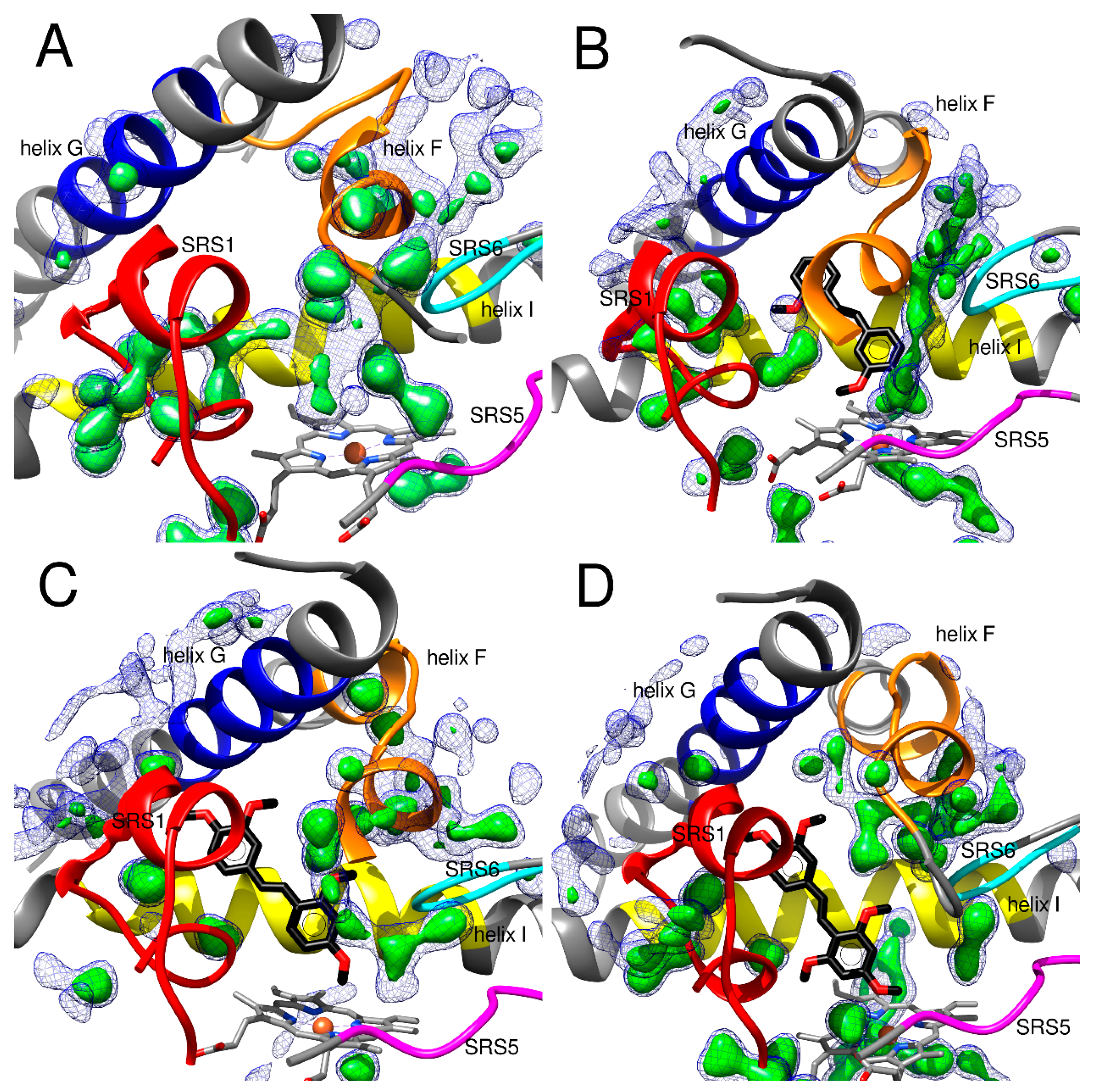
2.2.2. Hydration of CYP1A1 Binding Site
2.2.3. Hydration of the CYP1A2 Binding Site
2.2.4. Hydration of the CYP1B1 Binding Site
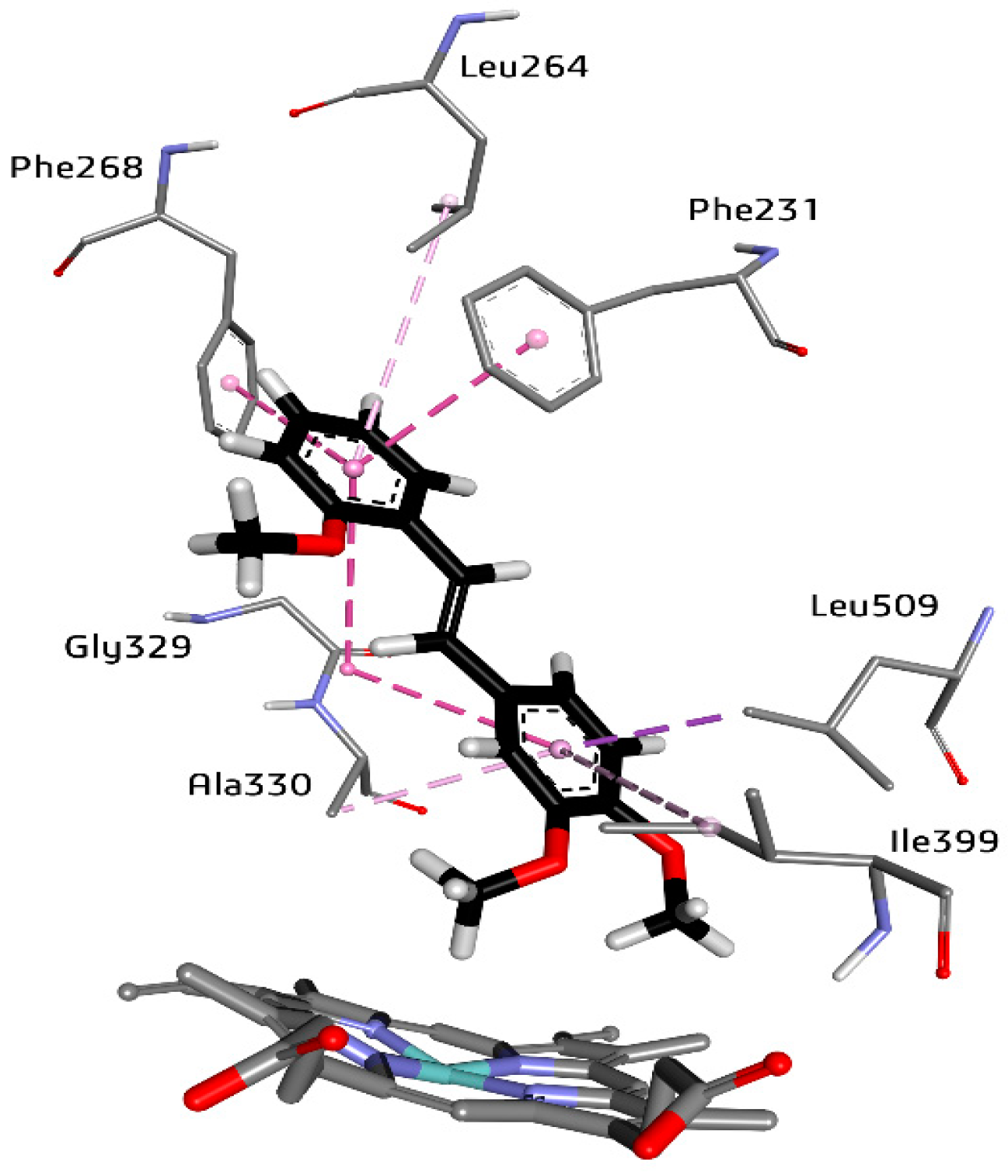
2.3. The High Affinity of 3,4,2′-triMS for the CYP1B1 Active Site
3. Materials and Methods
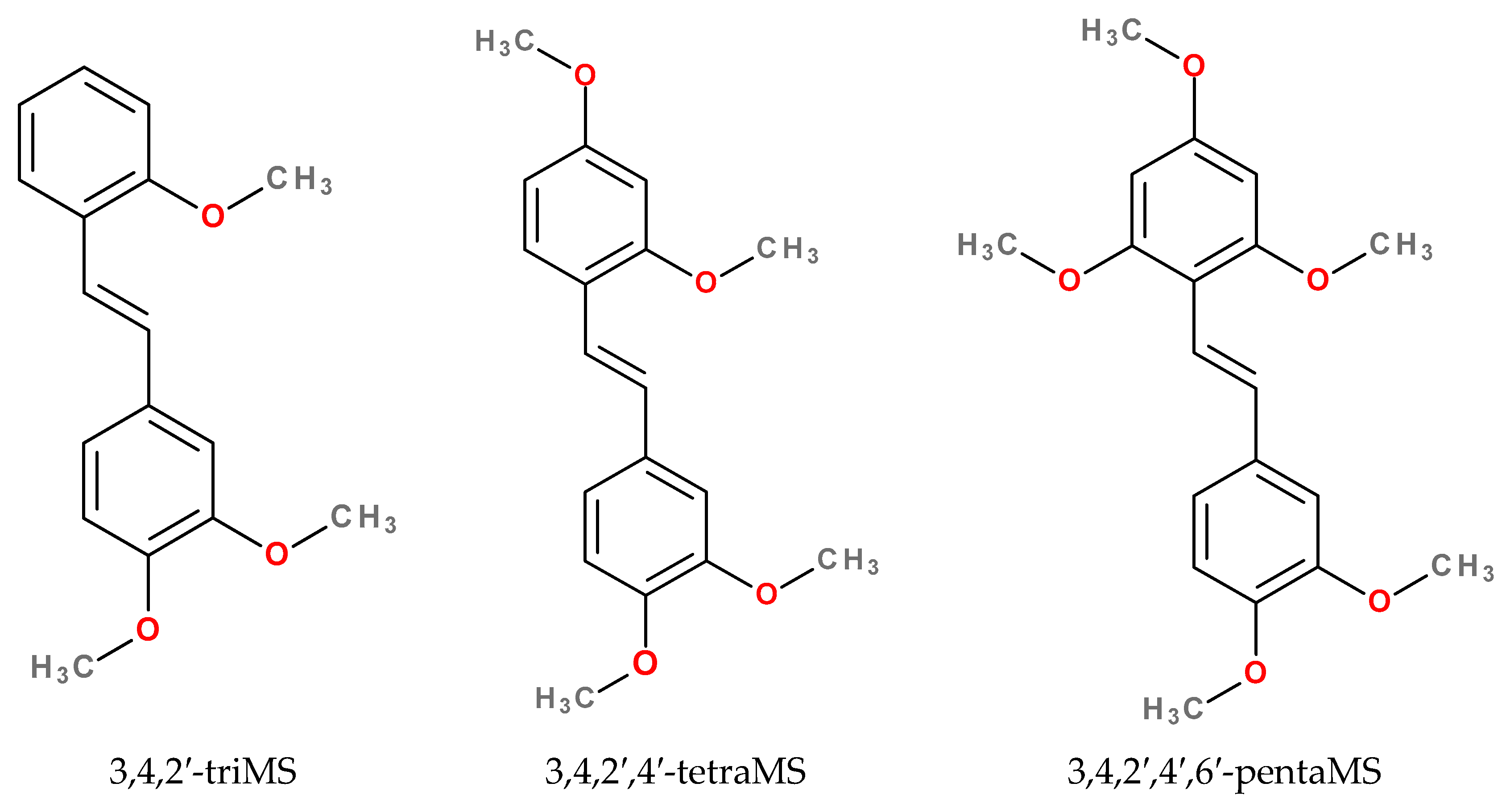
3.1. Selection of Ligands and Protein Structures
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
3.4. Tunnel Analysis
3.5. Averaging Protein Structures, Structural Changes Induced by Ligands, Hydrogen Bond Analysis, Water Molecule Occupancies, and Radius of Gyration of the Binding Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Presa, D.; Khurram, S.A.; Zubir, A.Z.A.; Smarakan, S.; Cooper, P.A.; Morais, G.R.; Sadiq, M.; Sutherland, M.; Loadman, P.M.; McCaul, J.; et al. Cytochrome P450 isoforms 1A1, 1B1 and 2W1 as targets for therapeutic intervention in head and neck cancer. Sci. Rep. 2021, 11, 18930. [Google Scholar] [CrossRef] [PubMed]
- Go, R.-E.; Hwang, K.-A.; Choi, K.-C. Cytochrome P450 1 family and cancers. J. Steroid Biochem. Mol. Biol. 2015, 147, 24–30. [Google Scholar] [CrossRef]
- Mikstacka, R.; Dutkiewicz, Z. New perspectives of CYP1B1 inhibitors in the light of molecular studies. Processes 2021, 9, 817. [Google Scholar] [CrossRef]
- Dutkiewicz, Z.; Mikstacka, R. Structure-based drug design for cytochrome P450 family 1 inhibitors. Bioinorg. Chem. Appl. 2018, 2018, 3924608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Itoh, T.; Yamamoto, K.; Takemura, H.; Shimoi, K. A 3D model of CYP1B1 explains the dominant 4-hydroxylation of estradiol. J. Chem. Inf. Model. 2010, 50, 1173–1178. [Google Scholar] [CrossRef]
- Alzahrani, A.M.; Rajendran, P. The multifarious link between cytochrome P450s and cancer. Oxidative Med. Cell. Longev. 2020, 2020, 3028387. [Google Scholar] [CrossRef]
- Akinwumi, B.C.; Bordun, K.-A.M.; Anderson, H.D. Biological activities of stilbenoids. Int. J. Mol. Sci. 2018, 19, 792. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Savas, U.; Stout, C.D.; Johnson, E.F. Structural characterization of the complex between α-naphthoflavone and human cytochrome P450 1B1. J. Biol. Chem. 2011, 286, 5736–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O. Cytochrome P450 structure-function: Insights from molecular dynamics simulations. Drug Metab. Rev. 2016, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P40 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef] [Green Version]
- Bart, A.G.; Takahashi, R.H.; Wang, X.; Scott, E.E. Human cytochrome P450 1A1 adapts active site for atypical nonplanar substrate. Drug Metab. Dispos. 2020, 48, 86–92. [Google Scholar] [CrossRef]
- Bart, A.G.; Morais, G.; Vangala, V.R.; Loadman, P.M.; Pors, K.; Scott, E.E. Cytochrome P450 binding and bioactivation of tumor-targeted duocarmycin agents. Drug. Metab. Dispos. 2022, 50, 49–57. [Google Scholar] [CrossRef]
- Kubo, M.; Yamamoto, K.; Itoh, T. Design and synthesis of selective CYP1B1 inhibitor via dearomatization of α-naphthoflavone. Bioorg. Med. Chem. 2019, 27, 285–304. [Google Scholar] [CrossRef]
- Bart, A.G.; Harris, K.L.; Gillam, E.M.J.; Scott, E.E. Structure of an ancestral mammalian family 1B1 cytochrome P450 with increased thermostability. J. Biol. Chem. 2020, 295, 5640–5653. [Google Scholar] [CrossRef] [Green Version]
- Juvonen, R.O.; Ahinko, M.; Jokinen, E.M.; Huuskonen, J.; Raunio, H.; Pentikainen, O.T. Substrate selectivity of coumarin derivatives by human CYP1 enzymes: In vitro enzyme kinetics and in silico modelling. ACS Omega 2021, 6, 11286–11296. [Google Scholar] [CrossRef]
- Wierzchowski, M.; Dutkiewicz, Z.; Gielara-Korzyńska, A.; Korzański, A.; Teubert, A.; Teżyk, A.; Stefański, T.; Baer-Dubowska, W.; Mikstacka, R. Synthesis, biological evaluation and docking studies of trans-stilbene methylthio derivatives as cytochromes P450 family 1 inhibitors. Chem. Biol. Drug Des. 2017, 90, 1226–1236. [Google Scholar] [CrossRef]
- Mikstacka, R.; Rimando, A.M.; Dutkiewicz, Z.; Stefański, T.; Sobiak, S. Design, synthesis and evaluation of the inhibitory selectivity of novel trans-resveratrol analogues on human recombinant CYP1A1, CYP1A2 and CYP1B1. Bioorg. Med. Chem. 2012, 20, 5117–5126. [Google Scholar] [CrossRef] [PubMed]
- Mikstacka, R.; Wierzchowski, M.; Dutkiewicz, Z.; Gielara-Korzańska, A.; Korzański, A.; Teubert, A.; Sobiak, S.; Baer-Dubowska, W. 3,4,2′-Trimethoxy-trans-stilbene—A potent CYP1B1 inhibitor. Med. Chem. Commun. 2014, 5, 496–501. [Google Scholar] [CrossRef]
- Chun, Y.J.; Lim, C.; Ohk, S.O.; Lee, J.M.; Lee, J.H.; Choi, S.; Kim, S. trans-Stilbenoids: Potent and selective inhibitors for human cytochrome P450 1B1. Med. Chem. Commun. 2011, 2, 402–405. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cho, H.; Thangapandian, S.; Lim, C.; Chun, Y.J.; Lee, Y.; Choi, S.; Kim, S. Adaptable small ligand of CYP1 enzymes for use in understanding the structural features determining isoform selectivity. ACS Med. Chem. Lett. 2018, 9, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Takemura, H.; Itoh, T.; Yamamoto, K.; Sakakibara, H.; Shimoi, K. Selective inhibition of methoxyflavonoids on human CYP1B1 activity. Bioorg. Med. Chem. 2010, 18, 6310–6315. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, Q.; Cui, Q.; Huang, G.; Pan, X.; Li, S. Flavonoids and naphthoflavonoids: Wider roles in the modulation of cytochrome P450 family 1 enzymes. ChemMedChem 2016, 11, 2102–2118. [Google Scholar] [CrossRef]
- Dong, J.; Wang, Z.; Cui, J.; Meng, Q.; Li, S. Synthesis and structure-activity relationship studies of α-naphthoflavone derivatives as CYP1B1 inhibitors. Eur. J. Med. Chem. 2020, 187, 111938. [Google Scholar] [CrossRef]
- Dong, J.; Huang, G.; Cui, Q.; Meng, Q.; Li, S.; Cui, J. Discovery of heterocycle-containing α-naphthoflavone derivatives as water-soluble, highly potent and selective CYP1B1 inhibitors. Eur. J. Med. Chem. 2021, 209, 112895. [Google Scholar] [CrossRef]
- Kesharwani, S.S.; Nandekar, P.P.; Pragyan, P.; Rathod, V.; Sangamwar, A.T. Characterization of differences in substrate specificity among CYP1A1, CYP1A2 and CYP1B1: An integral approach employing molecular docking and molecular dynamics simulations. J. Mol. Recognit. 2016, 29, 370–390. [Google Scholar] [CrossRef]
- Sridhar, J.; Goyal, N.; Liu, J.; Foroozesh, M. Review of ligand specific factors for CYP1A subfamily enzymes from molecular modelling studies reported to-date. Molecules 2017, 22, 1143. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Cojocaru, V.; Wade, R.C. Conformational diversity and ligand tunnels of mammalian cytochrome P450s. Biotechnol. Appl. Biochem. 2013, 60, 45–134. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, S.S.; Nandekar, P.P.; Pragyan, P.; Sangamwar, A.T. Comparative Proteomics among cytochrome P450 family 1 for differential substrate specificity. Protein J. 2014, 33, 536–548. [Google Scholar] [CrossRef]
- Biedermannova, L.; Schneider, B. Hydration of proteins and nucleic acids: Advances in experimental and theory. A review. Biochim. Biophys. Acta 2016, 1860, 1821–1835. [Google Scholar] [CrossRef] [PubMed]
- Zsidó, B.Z.; Hetényi, C. The role of water in ligand binding. Curr. Opin. Struct. Biol. 2021, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hendrychova, T.; Berka, K.; Navratilova, V.; Anzenbacher, P.; Otyepka, M. Dynamics and hydratation of the active sites of mammalian cytochromes P450 probed by molecular dynamics simulation. Curr. Drug Metab. 2012, 13, 177–189. [Google Scholar] [CrossRef]
- Swigonska, S.; Molcan, T.; Nynca, A.; Ciereszko, R.E. The involvement of CYP1A2 in biodegradation of dioxins in pigs. PLoS ONE 2022, 17, e0267162. [Google Scholar] [CrossRef]
- Geschwindner, S.; Ulander, J. The current impact of water thermodynamics for small-molecule drug discovery. Expert Opin. Drug Discov. 2019, 14, 1221–1225. [Google Scholar] [CrossRef]
- Watanabe, Y.; Fukuyoshi, S.; Kato, K.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Gouda, H.; Oda, A. Investigation of substrate recognition for cytochrome P450 1A2 mediated by water molecules using docking and molecular dynamics simulations. J. Mol. Graph. Model. 2017, 44, 325–336. [Google Scholar] [CrossRef]
- Rudling, A.; Orro, A.; Carlsson, J. Prediction of ordered water molecules in protein binding sites from molecular dynamics simulations: The impact of ligand binding on hydration networks. J. Chem. Inf. Model. 2018, 58, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER–A CHARMM-based MD docking algorithm. J. Comp. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes. Discovery Studio Release 2016; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Spassov, V.Z.; Flook, P.K.; Yan, L. LOOPER: A molecular mechanics-based algorithm for protein loop prediction. Protein Eng. Des. Sel. 2008, 21, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Ribeiro, J.V.; Bernardi, R.C.; Rudack, T.; Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Schulten, K. QWIKMD—integrative molecular dynamics toolkit for novices and experts. Sci. Rep. 2016, 6, 26536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. Charmm General Force Field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddart, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comp. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chovancová, E.; Pavelka, A.; Beneš, P.; Strnad, O.; Brezovský, J.; Kozlíková, B.; Gora, A.; Šustr, V.; Klvaňa, M.; Medek, P.; et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta 2007, 1770, 390–401. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (95% Confidence Interval) (μM) | Ratio | |||
|---|---|---|---|---|---|
| CYP1A1 | CYP1A2 | CYP1B1 | CYP1A1/CYP1B1 | CYP1A2/CYP1B1 | |
| 3,4,2′-triMS | 0.36 (0.27–0.48) | 4.93 (4.43–5.50) | 0.0034 (0.0016–0.0072) | 90 | 830 |
| 3,4,2′,4′-tetraMS | 0.51 (0.24–0.78) | >100 | 0.21 (0.14–0.28) | 2.4 | >480 |
| 3,4,2′,4′,6′-pentaMS | 0.27 (0.18–0.36) | >100 | 0.30 (0.25–0.35) | 0.9 | >333 |
| ANF * | 0.01 | 0.0038 | 0.0013 | 7.7 | 2.9 |
| Ligand | CYP1A1 | CYP1A2 | CYP1B1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Amino Acid | Substituent | Occupancy % | Amino Acid | Substituent | Occupancy % | Amino Acid | Substituent | Occupancy % | |
| 3,4,2′-triMS | Ser122 | 3-methoxy | 0.26 | Asn257 | 4-methoxy | 10.74 | – | – | – |
| 3,4,2′,4′-tetraMS | – | – | – | – | – | – | Ile399 | 4-methoxy | 0.02 |
| 3,4,2′,4′,6′-pentaMS | Asn255 Asn255 Ser122 | 4-methoxy 3-methoxy 2′-methoxy | 27.32 0.04 0.02 | Thr124 | 3-methoxy | 0.40 | Thr334 | 4-methoxy | 0.10 |
| Structure/Complex | CYP1A1 | CYP1A2 | CYP1B1 |
|---|---|---|---|
| X-ray | 8.53 | 8.41 | 8.65 |
| APO | 8.86 (+4%) | 9.12 (+8%) | 9.09 (+5%) |
| 3,4,2′-triMS | 9.11 (+7%) | 9.19 (+9%) | 8.93 (+3%) |
| 3,4,2′,4′-tetraMS | 8.85 (+4%) | 8.91 (+6%) | 9.06 (+5%) |
| 3,4,2′,4′,6′-pentaMS | 9.10 (+7%) | 9.51 (+13%) | 9.31 (+8%) |
| Ligand | Number of Water Molecules | |||||
|---|---|---|---|---|---|---|
| CYP1A1 | CYP1A2 | CYP1B1 | ||||
| 3.4 Å | 5.0 Å | 3.4 Å | 5.0 Å | 3.4 Å | 5.0 Å | |
| 3,4,2′-triMS | 4.7 | 8.5 | 5.1 | 10.5 | 3.6 | 5.7 |
| 3,4,2′,4′-tetraMS | 4.4 | 8.5 | 3.5 | 5.8 | 6.5 | 9.0 |
| 3,4,2′,4′,6′-pentaMS | 5.3 | 9.7 | 8.0 | 15.9 | 5.4 | 10.5 |
| Ligand | Substituent | H-Bond Occupancy (%) | |||||
|---|---|---|---|---|---|---|---|
| CYP1A1 | CYP1A2 | CYP1B1 | |||||
| Lig-Wat | Lig-Wat-Prot | Lig-Wat | Lig-Wat-Prot | Lig-Wat | Lig-Wat-Prot | ||
| 3,4,2′-triMS | 3-methoxy | 7 | 21 | 8 (Asn257) | 0 | ||
| 4-methoxy | 41 | 2 | 0 | ||||
| 2′-methoxy | 43 | 2 (Ser122) 15 (Asp313) | 36 | 8 (Gly316) | 0 | ||
| 3,4,2′,4′-tetraMS | 3-methoxy | 39 | 1 (Asn257) 6 (Asn312) | 1 | |||
| 4-methoxy | 2 | 26 | 11 (Asn257) | 38 | 2 (Thr334) 5 (Ile399) 12 (Leu509) | ||
| 2′-methoxy | 49 | 27 (Gly316) | 3 | ||||
| 4′-methoxy | 4 | 4 | 49 | ||||
| 3,4,2′,4′,6′-pentaMS | 3-methoxy | 11 | 3 (Asn255) | 15 | 2 (Ile386) | 17 | 7 (Ile399) |
| 4-methoxy | 13 | 23 | 1 (Ile399) | ||||
| 2′-methoxy | 28 | 4 (Ser120) 16 (Ser122) 8 (Asp313) | 39 | ||||
| 4′-methoxy | 18 | 4 (Ser122) | |||||
| 6′-methoxy | 3 | 23 | 6 (Asp320) | 41 | 6 (Gly329) 17 (Gln332) | ||
| 2a | 2b | 2c | 2ac | 2e | 2f | 3 | 4 | S/S1 | S2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| CYP1A1 | ||||||||||
| APO | 7% | 15% | 4% | 4% | ||||||
| 3,4,2′-triMS | 7% | 70% | 5% | 39% | 3% | 18% | 12% | |||
| 3,4,2′,4′-tetraMS | 2% | 67% | 11% | 4% | ||||||
| 3,4,2′,4′,6′-pentaMS | 2% | 51% | ||||||||
| CYP1A2 | ||||||||||
| APO | 8% | 25% | 1% | 1% | 4% | 1% | 5% | 23% | ||
| 3,4,2′-triMS | 1% | 17% | 5% | 5% | 1% | 2% | 10% | 60% | ||
| 3,4,2′,4′-tetraMS | 36% | 1% | 5% | |||||||
| 3,4,2′,4′,6′-pentaMS | 3% | 30% | 3% | 9% | 2% | 1% | 34% | |||
| CYP1B1 | ||||||||||
| APO | 20% | 25% | 3% | 3% | 10% | |||||
| 3,4,2′-triMS | 3% | 15% | 8% | 65% | 9% | |||||
| 3,4,2′,4′-tetraMS | 31% | 3% | 1% | 9% | ||||||
| 3,4,2′,4′,6′-pentaMS | 27% | 1% | 49% | 27% |
| Structure/Complex | CYP1A1 | CYP1A2 | CYP1B1 |
|---|---|---|---|
| X-ray | 10 (100%) | 32 (100%) | 13 (100%) |
| APO | 8 (80%) | 28 (87%) | 11 (85%) |
| 3,4,2′-triMS | 9 (90%) | 25 (78%) | 11 (85%) |
| 3,4,2′,4′-tetraMS | 8 (80%) | 25 (78%) | 11 (85%) |
| 3,4,2′,4′,6′-pentaMS | 8 (80%) | 24 (75%) | 12 (92%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutkiewicz, Z.; Mikstacka, R. Hydration and Structural Adaptations of the Human CYP1A1, CYP1A2, and CYP1B1 Active Sites by Molecular Dynamics Simulations. Int. J. Mol. Sci. 2023, 24, 11481. https://doi.org/10.3390/ijms241411481
Dutkiewicz Z, Mikstacka R. Hydration and Structural Adaptations of the Human CYP1A1, CYP1A2, and CYP1B1 Active Sites by Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2023; 24(14):11481. https://doi.org/10.3390/ijms241411481
Chicago/Turabian StyleDutkiewicz, Zbigniew, and Renata Mikstacka. 2023. "Hydration and Structural Adaptations of the Human CYP1A1, CYP1A2, and CYP1B1 Active Sites by Molecular Dynamics Simulations" International Journal of Molecular Sciences 24, no. 14: 11481. https://doi.org/10.3390/ijms241411481