
Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
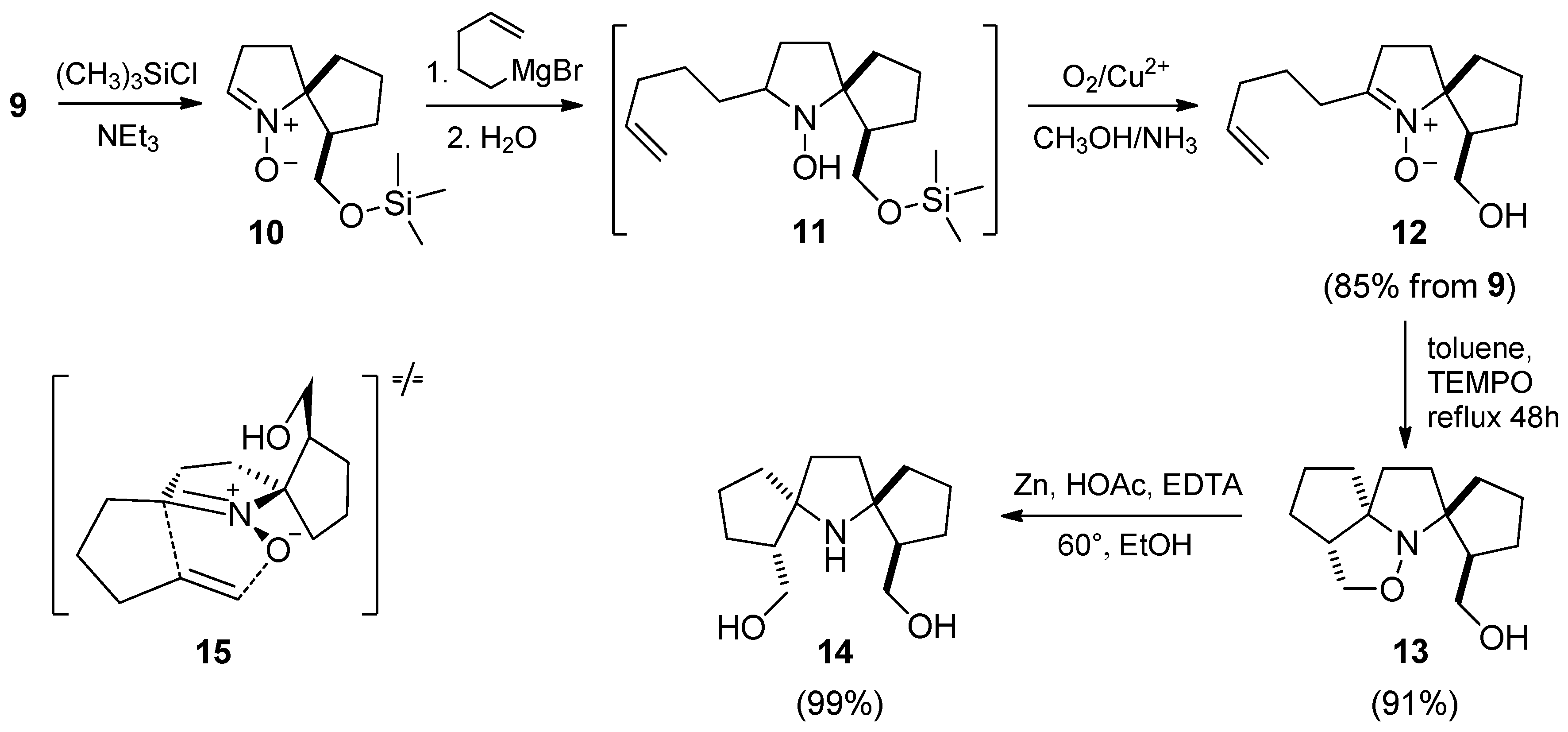{kind=link}
{kind=link}
{kind=link}
{kind=link}
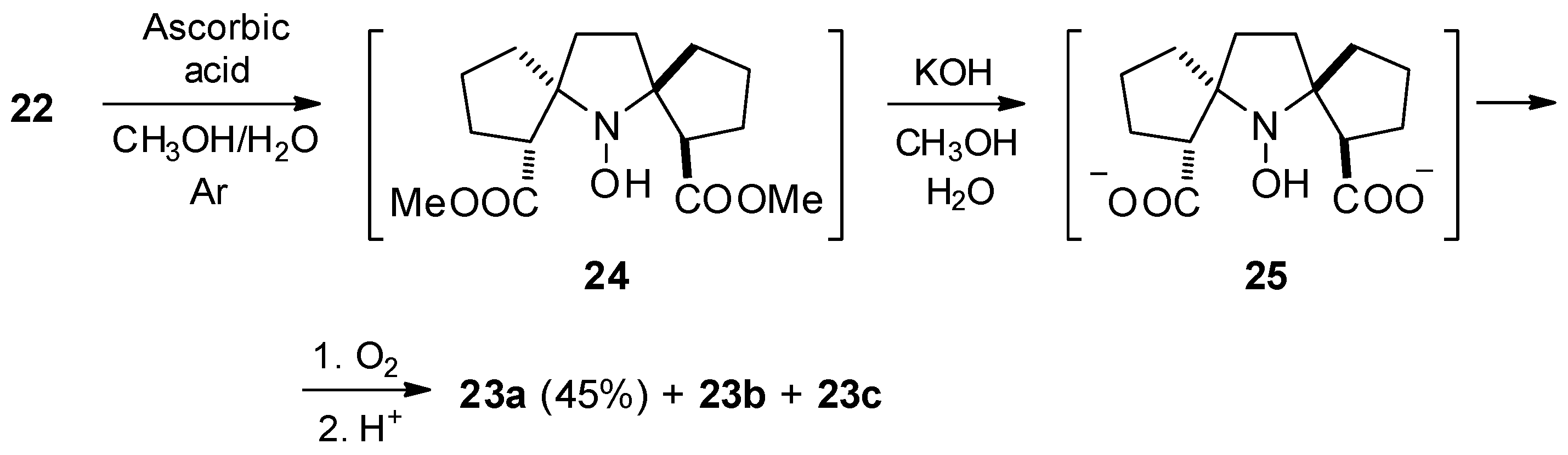{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Khoroshunova, Y.V.; Morozov, D.A.; Kuznetsov, D.A.; Rybalova, T.V.; Glazachev, Y.I.; Bagryanskaya, E.G.; Kirilyuk, I.A. Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times. Int. J. Mol. Sci. 2023, 24, 11498. https://doi.org/10.3390/ijms241411498
Khoroshunova YV, Morozov DA, Kuznetsov DA, Rybalova TV, Glazachev YI, Bagryanskaya EG, Kirilyuk IA. Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times. International Journal of Molecular Sciences. 2023; 24(14):11498. https://doi.org/10.3390/ijms241411498
Chicago/Turabian StyleKhoroshunova, Yulia V., Denis A. Morozov, Danil A. Kuznetsov, Tatyana V. Rybalova, Yurii I. Glazachev, Elena G. Bagryanskaya, and Igor A. Kirilyuk. 2023. "Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times" International Journal of Molecular Sciences 24, no. 14: 11498. https://doi.org/10.3390/ijms241411498
APA StyleKhoroshunova, Y. V., Morozov, D. A., Kuznetsov, D. A., Rybalova, T. V., Glazachev, Y. I., Bagryanskaya, E. G., & Kirilyuk, I. A. (2023). Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times. International Journal of Molecular Sciences, 24(14), 11498. https://doi.org/10.3390/ijms241411498