
Effects of Probiotics on Colitis-Induced Exacerbation of Alzheimer’s Disease in AppNL-G-F Mice
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Probiotic Ameliorated the DSS-Induced Colitis-like Condition in the Intestine
2.2. DSS-Induced Colitis and Probiotic Administration Increased Neutrophil Elastase Immunoreactivity in the Brain
2.3. Probiotic and DSS-Mediated Alterations in Brain Cytokines
2.4. DSS- and Probiotic-Mediated Alterations in Hippocampal Aβ Accumulation
2.5. DSS- and Probiotic-Associated Alterations in Hippocampal Gliosis
2.6. DSS- and Probiotic-Associated Alterations in c-Fos Immunoreactivity
2.7. DSS- and Probiotic-Associated Alterations in 5hmC Immunoreactivity
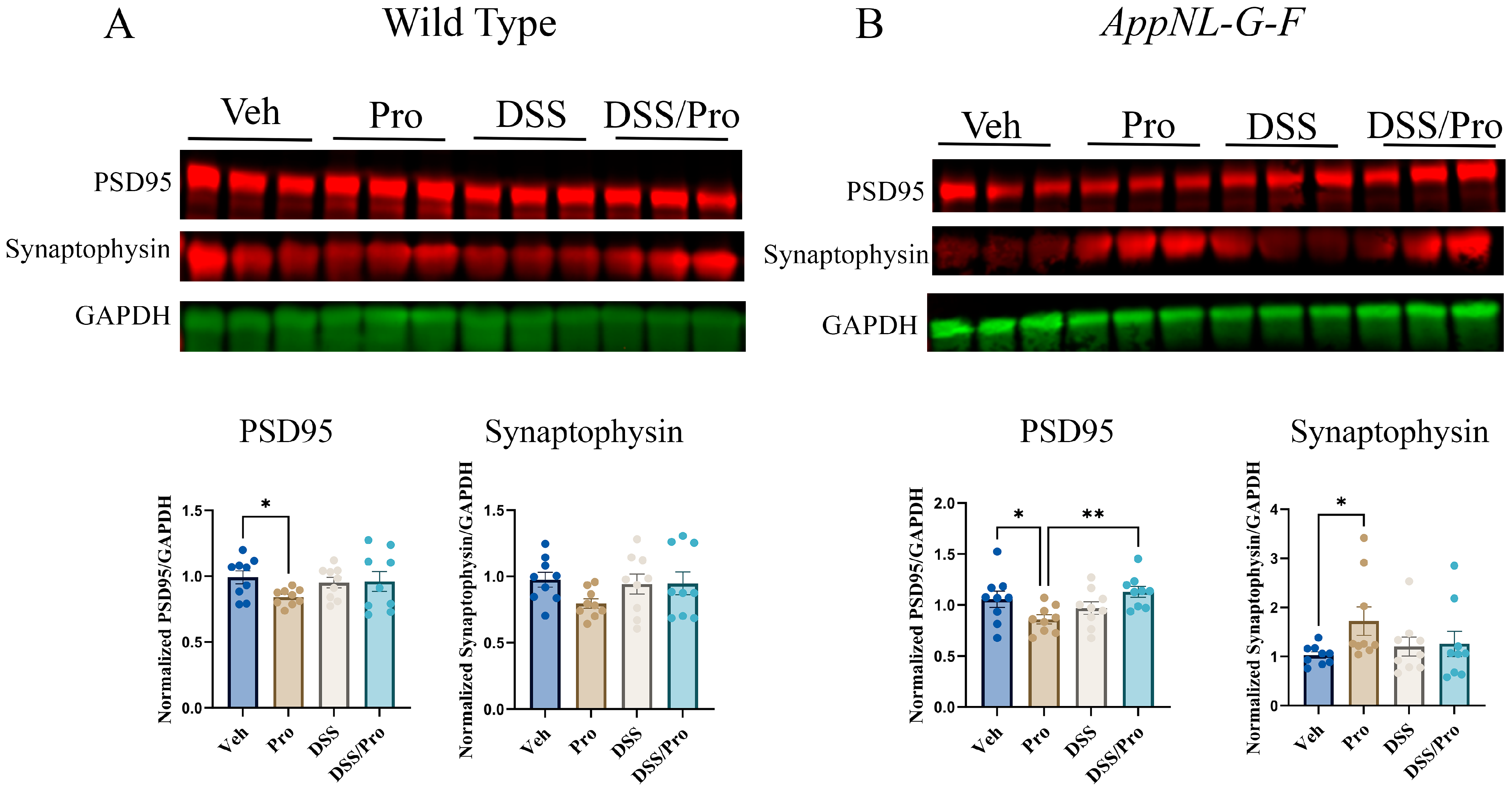
2.8. DSS- and Probiotic-Associated Alterations in Synaptic Proteins
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. DSS Exposure and Probiotic Treatment
4.3. Assessment of the Severity of Colitis-like Symptoms
4.4. Immunohistochemistry
4.4.1. Quantification of Aβ, GFAP, and Iba-1 Staining
4.4.2. Quantification of Elastase, c-Fos, and 5hmC Staining
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Western Blotting
4.7. Th1/Th2/Th17 Cytokine Assessment
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collaborators, G.B.D.D.F. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Huang, L.; Lu, Z.; Zhang, H.; Wen, H.; Li, Z.; Liu, Q.; Wang, R. A Novel Strategy for Alzheimer’s Disease Based on the Regulatory Effect of Amyloid-β on Gut Flora. J. Alzheimer’s Dis. JAD 2022, 1–13. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.P.; Meng, R.; Sun, Y.X.; Sun, W.J.; Ji, X.M.; Jia, L.F. Cerebrospinal fluid tau, Abeta1-42 and inflammatory cytokines in patients with Alzheimer’s disease and vascular dementia. Neurosci. Lett. 2005, 383, 12–16. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Welge, V.; Fiege, O.; Lewczuk, P.; Mollenhauer, B.; Esselmann, H.; Klafki, H.W.; Wolf, S.; Trenkwalder, C.; Otto, M.; Kornhuber, J.; et al. Combined CSF tau, p-tau181 and amyloid-beta 38/40/42 for diagnosing Alzheimer’s disease. J. Neural Transm. 2009, 116, 203–212. [Google Scholar] [CrossRef]
- Dos Santos Picanco, L.C.; Ozela, P.F.; de Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; de Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimers Dis. 2022, 87, 83–100. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of Aducanumab in the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef]
- Boeri, L.; Perottoni, S.; Izzo, L.; Giordano, C.; Albani, D. Microbiota-Host Immunity Communication in Neurodegenerative Disorders: Bioengineering Challenges for In Vitro Modeling. Adv. Health Mater. 2021, 10, e2002043. [Google Scholar] [CrossRef]
- Junges, V.M.; Closs, V.E.; Nogueira, G.M.; Gottlieb, M.G.V. Crosstalk between Gut Microbiota and Central Nervous System: A Focus on Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 1179–1190. [Google Scholar] [CrossRef]
- Kaur, H.; Singh, Y.; Singh, S.; Singh, R.B. Gut microbiome-mediated epigenetic regulation of brain disorder and application of machine learning for multi-omics data analysis. Genome 2021, 64, 355–371. [Google Scholar] [CrossRef]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Luan, H.; Wang, X.; Cai, Z. Mass spectrometry-based metabolomics: Targeting the crosstalk between gut microbiota and brain in neurodegenerative disorders. Mass. Spectrom. Rev. 2019, 38, 22–33. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Gut instincts: Microbiota as a key regulator of brain development, ageing and neurodegeneration. J. Physiol. 2017, 595, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Corn, G.; Shrestha, S.; Nielsen, N.M.; Frisch, M.; Colombel, J.F.; Jess, T. Inflammatory bowel diseases among first-generation and second-generation immigrants in Denmark: A population-based cohort study. Gut 2021, 70, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Hamilton, M.J. The valuable role of endoscopy in inflammatory bowel disease. Diagn. Ther. Endosc. 2012, 2012, 467979. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Jiang, Y.; Xu, K.; Cui, M.; Ye, W.; Zhao, G.; Jin, L.; Chen, X. The progress of gut microbiome research related to brain disorders. J. Neuroinflam. 2020, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gao, M.; Liu, Z.; Zhang, Y.; Tu, H.; Lei, L.; Wu, P.; Zhang, A.; Yang, C.; Li, G.; et al. Gut Microbiome Composition Linked to Inflammatory Factors and Cognitive Functions in First-Episode, Drug-Naive Major Depressive Disorder Patients. Front. Neurosci. 2021, 15, 800764. [Google Scholar] [CrossRef]
- Solas, M.; Milagro, F.I.; Ramirez, M.J.; Martinez, J.A. Inflammation and gut-brain axis link obesity to cognitive dysfunction: Plausible pharmacological interventions. Curr. Opin. Pharmacol. 2017, 37, 87–92. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Carna, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 78, 683–697. [Google Scholar] [CrossRef]
- Aggarwal, M.; Alkhayyat, M.; Abou Saleh, M.; Sarmini, M.T.; Singh, A.; Garg, R.; Garg, P.; Mansoor, E.; Padival, R.; Cohen, B.L. Alzheimer Disease Occurs More Frequently In Patients With Inflammatory Bowel Disease: Insight From a Nationwide Study. J. Clin. Gastroenterol. 2022, 57, 501–507. [Google Scholar] [CrossRef]
- Adewuyi, E.O.; O’Brien, E.K.; Nyholt, D.R.; Porter, T.; Laws, S.M. A large-scale genome-wide cross-trait analysis reveals shared genetic architecture between Alzheimer’s disease and gastrointestinal tract disorders. Commun. Biol. 2022, 5, 691. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Wiatrak, B.; Szelag, A. The Risk of Developing Alzheimer’s Disease and Parkinson’s Disease in Patients with Inflammatory Bowel Disease: A Meta-Analysis. J. Clin. Med. 2022, 11, 3704. [Google Scholar] [CrossRef]
- Sohrabi, M.; Pecoraro, H.L.; Combs, C.K. Gut Inflammation Induced by Dextran Sulfate Sodium Exacerbates Amyloid-beta Plaque Deposition in the AppNL-G-F Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 79, 1235–1255. [Google Scholar] [CrossRef]
- Kaur, H.; Golovko, S.; Golovko, M.Y.; Singh, S.; Darland, D.C.; Combs, C.K. Effects of Probiotic Supplementation on Short Chain Fatty Acids in the AppNL-G-F Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 1083–1102. [Google Scholar] [CrossRef]
- Chassaing, B.; Srinivasan, G.; Delgado, M.A.; Young, A.N.; Gewirtz, A.T.; Vijay-Kumar, M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS ONE 2012, 7, e44328. [Google Scholar] [CrossRef] [Green Version]
- in t’ Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef] [Green Version]
- Nichols, M.R.; St-Pierre, M.K.; Wendeln, A.C.; Makoni, N.J.; Gouwens, L.K.; Garrad, E.C.; Sohrabi, M.; Neher, J.J.; Tremblay, M.E.; Combs, C.K. Inflammatory mechanisms in neurodegeneration. J. Neurochem. 2019, 149, 562–581. [Google Scholar] [CrossRef] [Green Version]
- Riazi, K.; Galic, M.A.; Kuzmiski, J.B.; Ho, W.; Sharkey, K.A.; Pittman, Q.J. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 17151–17156. [Google Scholar] [CrossRef]
- Roach, M.; Christie, J.A. Fecal incontinence in the elderly. Geriatrics 2008, 63, 13–22. [Google Scholar]
- Cai, L.; Li, X.; Geng, C.; Lei, X.; Wang, C. Molecular mechanisms of somatostatin-mediated intestinal epithelial barrier function restoration by upregulating claudin-4 in mice with DSS-induced colitis. Am. J. Physiol. Cell Physiol. 2018, 315, C527–C536. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.; Ruano-Gallego, D.; Radhakrishnan, S.T.; Lovell, S.; Yu, L.; Kotik, O.; Glegola-Madejska, I.; Tate, E.W.; Choudhary, J.S.; Williams, H.R.T.; et al. Faecal neutrophil elastase-antiprotease balance reflects colitis severity. Mucosal Immunol. 2020, 13, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumbhare, D.; Palys, V.; Toms, J.; Wickramasinghe, C.S.; Amarasinghe, K.; Manic, M.; Hughes, E.; Holloway, K.L. Nucleus Basalis of Meynert Stimulation for Dementia: Theoretical and Technical Considerations. Front. Neurosci. 2018, 12, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Ma, Q.; Miao, L.; Yang, H.; Pan, L.; Li, K.; Zeng, L.H.; Zhang, X.; Wu, J.; Hao, S.; et al. A substantia innominata-midbrain circuit controls a general aggressive response. Neuron 2021, 109, 1540–1553.e1549. [Google Scholar] [CrossRef]
- Poli, V.; Zanoni, I. Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease. Trends Microbiol. 2023, 31, 280–293. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafo, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca(2+) Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.U.; Anwar, L.; Ali, M.H.; Iqubal, M.K.; Iqubal, A.; Baboota, S.; Ali, J. Signalling Pathways Involved in Microglial Activation in Alzheimer’s Disease and Potential Neuroprotective Role of Phytoconstituents. CNS Neurol. Disord. Drug Targets 2022. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Taipa, R.; Brochado, P.; Robinson, A.; Reis, I.; Costa, P.; Mann, D.M.; Melo Pires, M.; Sousa, N. Patterns of Microglial Cell Activation in Alzheimer Disease and Frontotemporal Lobar Degeneration. Neurodegener. Dis. 2017, 17, 145–154. [Google Scholar] [CrossRef]
- Walton, M.; MacGibbon, G.; Young, D.; Sirimanne, E.; Williams, C.; Gluckman, P.; Dragunow, M. Do c-Jun, c-Fos, and amyloid precursor protein play a role in neuronal death or survival? J. Neurosci. Res. 1998, 53, 330–342. [Google Scholar] [CrossRef]
- Yap, E.L.; Pettit, N.L.; Davis, C.P.; Nagy, M.A.; Harmin, D.A.; Golden, E.; Dagliyan, O.; Lin, C.; Rudolph, S.; Sharma, N.; et al. Bidirectional perisomatic inhibitory plasticity of a Fos neuronal network. Nature 2021, 590, 115–121. [Google Scholar] [CrossRef]
- Antunes, C.; Da Silva, J.D.; Guerra-Gomes, S.; Alves, N.D.; Ferreira, F.; Loureiro-Campos, E.; Branco, M.R.; Sousa, N.; Reik, W.; Pinto, L.; et al. Tet3 ablation in adult brain neurons increases anxiety-like behavior and regulates cognitive function in mice. Mol. Psychiatry 2021, 26, 1445–1457. [Google Scholar] [CrossRef]
- Greer, C.B.; Wright, J.; Weiss, J.D.; Lazarenko, R.M.; Moran, S.P.; Zhu, J.; Chronister, K.S.; Jin, A.Y.; Kennedy, A.J.; Sweatt, J.D.; et al. Tet1 Isoforms Differentially Regulate Gene Expression, Synaptic Transmission, and Memory in the Mammalian Brain. J. Neurosci. 2021, 41, 578–593. [Google Scholar] [CrossRef]
- Colquitt, B.M.; Allen, W.E.; Barnea, G.; Lomvardas, S. Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc. Natl. Acad. Sci. USA 2013, 110, 14682–14687. [Google Scholar] [CrossRef]
- Ishikawa, H.; Matsumoto, S.; Ohashi, Y.; Imaoka, A.; Setoyama, H.; Umesaki, Y.; Tanaka, R.; Otani, T. Beneficial effects of probiotic bifidobacterium and galacto-oligosaccharide in patients with ulcerative colitis: A randomized controlled study. Digestion 2011, 84, 128–133. [Google Scholar] [CrossRef]
- Palumbo, V.D.; Romeo, M.; Gammazza, A.M.; Carini, F.; Damiani, P.; Damiano, G.; Buscemi, S.; Monte, A.I.L.; Gerges-Geagea, A.; Jurjus, A. The long-term effects of probiotics in the therapy of ulcerative colitis: A clinical study. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2016, 160, 372–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L.; Wang, X.H.; Cui, Y.; Lian, G.H.; Zhang, J.; Ouyang, C.H.; Lu, F.G. Therapeutic effects of four strains of probiotics on experimental colitis in mice. World J. Gastroenterol. 2009, 15, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Wasilewski, A.; Zielińska, M.; Storr, M.; Fichna, J. Beneficial effects of probiotics, prebiotics, synbiotics, and psychobiotics in inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Vakadaris, G.; Stefanis, C.; Giorgi, E.; Brouvalis, M.; Voidarou, C.C.; Kourkoutas, Y.; Tsigalou, C.; Bezirtzoglou, E. The Role of Probiotics in Inducing and Maintaining Remission in Crohn’s Disease and Ulcerative Colitis: A Systematic Review of the Literature. Biomedicines 2023, 11, 494. [Google Scholar] [CrossRef]
- Martín, R.; Chain, F.; Miquel, S.; Motta, J.-P.; Vergnolle, N.; Sokol, H.; Langella, P. Using murine colitis models to analyze probiotics–host interactions. FEMS Microbiol. Rev. 2017, 41, S49–S70. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.; Kim, H.J. Intestinal barrier dysfunction orchestrates the onset of inflammatory host-microbiome cross-talk in a human gut inflammation-on-a-chip. Proc. Natl. Acad. Sci. USA 2018, 115, E10539–E10547. [Google Scholar] [CrossRef] [Green Version]
- Manocha, G.D.; Floden, A.M.; Miller, N.M.; Smith, A.J.; Nagamoto-Combs, K.; Saito, T.; Saido, T.C.; Combs, C.K. Temporal progression of Alzheimer’s disease in brains and intestines of transgenic mice. Neurobiol. Aging 2019, 81, 166–176. [Google Scholar] [CrossRef]
- Varesi, A.; Pierella, E.; Romeo, M.; Piccini, G.B.; Alfano, C.; Bjørklund, G.; Oppong, A.; Ricevuti, G.; Esposito, C.; Chirumbolo, S. The potential role of gut microbiota in Alzheimer’s disease: From diagnosis to treatment. Nutrients 2022, 14, 668. [Google Scholar] [CrossRef]
- Bostanciklioğlu, M. The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 2019, 127, 954–967. [Google Scholar] [CrossRef]
- Hu, X.; Wang, T.; Jin, F. Alzheimer’s disease and gut microbiota. Sci. China Life Sci. 2016, 59, 1006–1023. [Google Scholar] [CrossRef] [Green Version]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut microbiota and their neuroinflammatory implications in Alzheimer’s disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef] [Green Version]
- Goyal, D.; Ali, S.A.; Singh, R.K. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106, 110112. [Google Scholar] [CrossRef]
- Kanashiro, A.; Hiroki, C.H.; da Fonseca, D.M.; Birbrair, A.; Ferreira, R.G.; Bassi, G.S.; Fonseca, M.D.; Kusuda, R.; Cebinelli, G.C.M.; da Silva, K.P.; et al. The role of neutrophils in neuro-immune modulation. Pharmacol. Res. 2020, 151, 104580. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Stock, A.J.; Kasus-Jacobi, A.; Pereira, H.A. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J. Neuroinflam. 2018, 15, 240. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, R.; Bastos, P.A.D.; Disson, O.; Rifflet, A.; Gabanyi, I.; Spielbauer, J.; Berard, M.; Lecuit, M.; Boneca, I.G. Microbiota-induced active translocation of peptidoglycan across the intestinal barrier dictates its within-host dissemination. Proc. Natl. Acad. Sci. USA 2023, 120, e2209936120. [Google Scholar] [CrossRef]
- Teipel, S.J.; Grinberg, L.T.; Hampel, H.; Heinsen, H. Cholinergic System Imaging in the Healthy Aging Process and Alzheimer Disease. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009; pp. 857–868. [Google Scholar] [CrossRef]
- Parada, E.; Egea, J.; Buendia, I.; Negredo, P.; Cunha, A.C.; Cardoso, S.; Soares, M.P.; Lopez, M.G. The microglial alpha7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid. Redox Signal 2013, 19, 1135–1148. [Google Scholar] [CrossRef] [Green Version]
- Revathikumar, P.; Bergqvist, F.; Gopalakrishnan, S.; Korotkova, M.; Jakobsson, P.J.; Lampa, J.; Le Maitre, E. Immunomodulatory effects of nicotine on interleukin 1beta activated human astrocytes and the role of cyclooxygenase 2 in the underlying mechanism. J. Neuroinflam. 2016, 13, 256. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Li, X.; Hao, J. The cholinergic anti-inflammatory pathway: An innovative treatment strategy for neurological diseases. Neurosci. Biobehav. Rev. 2017, 77, 358–368. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex--linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef]
- Kaur, H.; Nagamoto-Combs, K.; Golovko, S.; Golovko, M.Y.; Klug, M.G.; Combs, C.K. Probiotics ameliorate intestinal pathophysiology in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 92, 114–134. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, G.; Combs, C.K. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J. Neuroinflam. 2012, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojanathammanee, L.; Floden, A.M.; Manocha, G.D.; Combs, C.K. Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J. Neuroinflam. 2015, 12, 42. [Google Scholar] [CrossRef] [Green Version]
- Frautschy, S.A.; Yang, F.; Irrizarry, M.; Hyman, B.; Saido, T.C.; Hsiao, K.; Cole, G.M. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 1998, 152, 307–317. [Google Scholar] [PubMed]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef]
- Stalder, M.; Phinney, A.; Probst, A.; Sommer, B.; Staufenbiel, M.; Jucker, M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 1999, 154, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Braak, H.; Del Tredici, K.; Leyh, J.; Lier, J.; Khoshbouei, H.; Eisenlöffel, C.; Müller, W.; Bechmann, I. Microglial activation occurs late during preclinical Alzheimer’s disease. Glia 2018, 66, 2550–2562. [Google Scholar] [CrossRef]
- Dickson, D.W.; Farlo, J.; Davies, P.; Crystal, H.; Fuld, P.; Yen, S.H. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am. J. Pathol. 1988, 132, 86–101. [Google Scholar]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Occurrence of HLA-DR reactive microglia in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1988, 540, 319–323. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Barron, E.; Chui, H.C. Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci. Lett. 1990, 119, 32–36. [Google Scholar] [CrossRef]
- Tooyama, I.; Kimura, H.; Akiyama, H.; McGeer, P.L. Reactive microglia express class I and class II major histocompatibility complex antigens in Alzheimer’s disease. Brain Res. 1990, 523, 273–280. [Google Scholar] [CrossRef]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.; El-Okl, M.; Williams, A.L.; Cunningham, C.; Wilcockson, D.; Perry, V.H. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 788–789. [Google Scholar] [CrossRef]
- Cai, H.; Liang, Q.; Ge, G. Gypenoside Attenuates beta Amyloid-Induced Inflammation in N9 Microglial Cells via SOCS1 Signaling. Neural Plast. 2016, 2016, 6362707. [Google Scholar] [CrossRef] [Green Version]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhao, R.; Gao, K.; Wei, Z.; Yin, M.Y.; Lau, L.T.; Chui, D.; Yu, A.C. Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Sajja, V.S.; Hlavac, N.; VandeVord, P.J. Role of Glia in Memory Deficits Following Traumatic Brain Injury: Biomarkers of Glia Dysfunction. Front. Integr. Neurosci. 2016, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Floden, A.M.; Combs, C.K. Beta-amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 4644–4648. [Google Scholar] [CrossRef] [Green Version]
- Floden, A.M.; Combs, C.K. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J. Alzheimer’s Dis. JAD 2011, 25, 279–293. [Google Scholar] [CrossRef]
- Floden, A.M.; Li, S.; Combs, C.K. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 2566–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornemann, K.D.; Wiederhold, K.H.; Pauli, C.; Ermini, F.; Stalder, M.; Schnell, L.; Sommer, B.; Jucker, M.; Staufenbiel, M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am. J. Pathol. 2001, 158, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 5709–5714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lue, L.F.; Walker, D.G.; Rogers, J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol. Aging 2001, 22, 945–956. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Picciano, M.; Malester, B.; LaFrancois, J.; Zehr, C.; Daeschner, J.M.; Olschowka, J.A.; Fonseca, M.I.; O’Banion, M.K.; Tenner, A.J.; et al. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 2001, 158, 1345–1354. [Google Scholar] [CrossRef]
- Monsonego, A.; Imitola, J.; Petrovic, S.; Zota, V.; Nemirovsky, A.; Baron, R.; Fisher, Y.; Owens, T.; Weiner, H.L. Abeta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5048–5053. [Google Scholar] [CrossRef]
- Townsend, K.P.; Town, T.; Mori, T.; Lue, L.F.; Shytle, D.; Sanberg, P.R.; Morgan, D.; Fernandez, F.; Flavell, R.A.; Tan, J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur. J. Immunol. 2005, 35, 901–910. [Google Scholar] [CrossRef]
- Wu, Q.; Combs, C.; Cannady, S.B.; Geldmacher, D.S.; Herrup, K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol. Aging 2000, 21, 797–806. [Google Scholar] [CrossRef]
- Pettit, N.L.; Yap, E.L.; Greenberg, M.E.; Harvey, C.D. Fos ensembles encode and shape stable spatial maps in the hippocampus. Nature 2022, 609, 327–334. [Google Scholar] [CrossRef]
- Cruz-Mendoza, F.; Jauregui-Huerta, F.; Aguilar-Delgadillo, A.; García-Estrada, J.; Luquin, S. Immediate Early Gene c-fos in the Brain: Focus on Glial Cells. Brain Sci. 2022, 12, 687. [Google Scholar] [CrossRef]
- Narita, M.; Imai, S.; Oe, K.; Narita, M.; Kubota, C.; Yajima, Y.; Yamazaki, M.; Suzuki, T. Induction of c-fos expression in the mouse brain associated with hyperalgesia induced by intrathecal injection of protein kinase C activator. Brain Res. 2004, 1015, 189–193. [Google Scholar] [CrossRef]
- Knezovic, A.; Budisa, S.; Babic Perhoc, A.; Homolak, J.; Osmanovic Barilar, J. From Determining Brain Insulin Resistance in a Sporadic Alzheimer’s Disease Model to Exploring the Region-Dependent Effect of Intranasal Insulin. Mol. Neurobiol. 2023, 60, 2005–2023. [Google Scholar] [CrossRef]
- Phothi, T.; Tunsophon, S.; Tiyaboonchai, W.; Khongsombat, O. Effects of curcumin and γ-oryzanol solid dispersion on the brain of middle-aged rats. Biomed. Rep. 2022, 17, 59. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, H.; Cheng, R.; Tang, Z.; Zhang, M. Outer membrane protein Amuc_1100 of Akkermansia muciniphila alleviates antibiotic-induced anxiety and depression-like behavior in mice. Physiol. Behav. 2023, 258, 114023. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, I.C.; Dawlaty, M.M. TET Enzymes and 5-Hydroxymethylcytosine in Neural Progenitor Cell Biology and Neurodevelopment. Front. Cell Dev. Biol. 2021, 9, 645335. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Li, L.; Xu, K.; Ma, Z.; Chow, H.-M.; Herrup, K.; Li, J. Selective loss of 5hmC promotes neurodegeneration in the mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 16364–16382. [Google Scholar] [CrossRef]
- Cuomo, M.; Borrelli, L.; Della Monica, R.; Coretti, L.; De Riso, G.; D’Angelo Lancellotti di Durazzo, L.; Fioretti, A.; Lembo, F.; Dinan, T.G.; Cryan, J.F.; et al. DNA Methylation Profiles of Tph1A and BDNF in Gut and Brain of L. Rhamnosus-Treated Zebrafish. Biomolecules 2021, 11, 142. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, T.; Xu, Y.; Gu, X.; Li, D.; Niu, K.; Wang, T.; Zhao, J.; Zhou, R.; Wang, H.L. Long-term probiotic intervention mitigates memory dysfunction through a novel H3K27me3-based mechanism in lead-exposed rats. Transl. Psychiatry 2020, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, 60, e3678. [Google Scholar] [CrossRef] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmora, N.; Zilberman-Schapira, G.; Suez, J.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Kotler, E.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.; et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell 2018, 174, 1388–1405.e1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Ghia, J.E.; Blennerhassett, P.; Kumar-Ondiveeran, H.; Verdu, E.F.; Collins, S.M. The vagus nerve: A tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006, 131, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Snider, A.J.; Bialkowska, A.B.; Ghaleb, A.M.; Yang, V.W.; Obeid, L.M.; Hannun, Y.A. Murine Model for Colitis-Associated Cancer of the Colon. Methods Mol. Biol. 2016, 1438, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Nagamoto-Combs, K.; Manocha, G.D.; Puig, K.; Combs, C.K. An improved approach to align and embed multiple brain samples in a gelatin-based matrix for simultaneous histological processing. J. Neurosci. Methods 2016, 261, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Morriss, N.J.; Conley, G.M.; Ospina, S.M.; Meehan Iii, W.P.; Qiu, J.; Mannix, R. Automated Quantification of Immunohistochemical Staining of Large Animal Brain Tissue Using QuPath Software. Neuroscience 2020, 429, 235–244. [Google Scholar] [CrossRef]
- Finney, C.A.; Jones, N.M.; Morris, M.J. A scalable, fully automated approach for regional quantification of immunohistochemical staining of astrocytes in the rat brain. J. Neurosci. Methods 2021, 348, 108994. [Google Scholar] [CrossRef]
- Hein, A.L.; Mukherjee, M.; Talmon, G.A.; Natarajan, S.K.; Nordgren, T.M.; Lyden, E.; Hanson, C.K.; Cox, J.L.; Santiago-Pintado, A.; Molani, M.A.; et al. QuPath Digital Immunohistochemical Analysis of Placental Tissue. J. Pathol. Inf. 2021, 12, 40. [Google Scholar] [CrossRef]
- Lee, A.; Jiang, Z.; Zhu, L.; Ladiges, W. QuPath. A new digital imaging tool for geropathology. Aging Pathobiol. Ther. 2020, 2, 114–116. [Google Scholar] [CrossRef]
- Ribeiro, G.P.; Endringer, D.C.; De Andrade, T.U.; Lenz, D. Comparison between two programs for image analysis, machine learning and subsequent classification. Tissue Cell 2019, 58, 12–16. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, B.; Johnson, L.M.; Sohrabi, M.; Usatii, A.A.; Craig, R.M.J.; Kaelberer, J.B.; Chandrasekaran, S.P.; Kaur, H.; Nookala, S.; Combs, C.K. Effects of Probiotics on Colitis-Induced Exacerbation of Alzheimer’s Disease in AppNL-G-F Mice. Int. J. Mol. Sci. 2023, 24, 11551. https://doi.org/10.3390/ijms241411551
Sahu B, Johnson LM, Sohrabi M, Usatii AA, Craig RMJ, Kaelberer JB, Chandrasekaran SP, Kaur H, Nookala S, Combs CK. Effects of Probiotics on Colitis-Induced Exacerbation of Alzheimer’s Disease in AppNL-G-F Mice. International Journal of Molecular Sciences. 2023; 24(14):11551. https://doi.org/10.3390/ijms241411551
Chicago/Turabian StyleSahu, Bijayani, Lauren M. Johnson, Mona Sohrabi, Anastasia A. Usatii, Rachel M. J. Craig, Joshua B. Kaelberer, Sathiya Priya Chandrasekaran, Harpreet Kaur, Suba Nookala, and Colin K. Combs. 2023. "Effects of Probiotics on Colitis-Induced Exacerbation of Alzheimer’s Disease in AppNL-G-F Mice" International Journal of Molecular Sciences 24, no. 14: 11551. https://doi.org/10.3390/ijms241411551
APA StyleSahu, B., Johnson, L. M., Sohrabi, M., Usatii, A. A., Craig, R. M. J., Kaelberer, J. B., Chandrasekaran, S. P., Kaur, H., Nookala, S., & Combs, C. K. (2023). Effects of Probiotics on Colitis-Induced Exacerbation of Alzheimer’s Disease in AppNL-G-F Mice. International Journal of Molecular Sciences, 24(14), 11551. https://doi.org/10.3390/ijms241411551