

Novel Cellular Functions of ATR for Therapeutic Targeting: Embryogenesis to Tumorigenesis

Abstract
1. Introduction
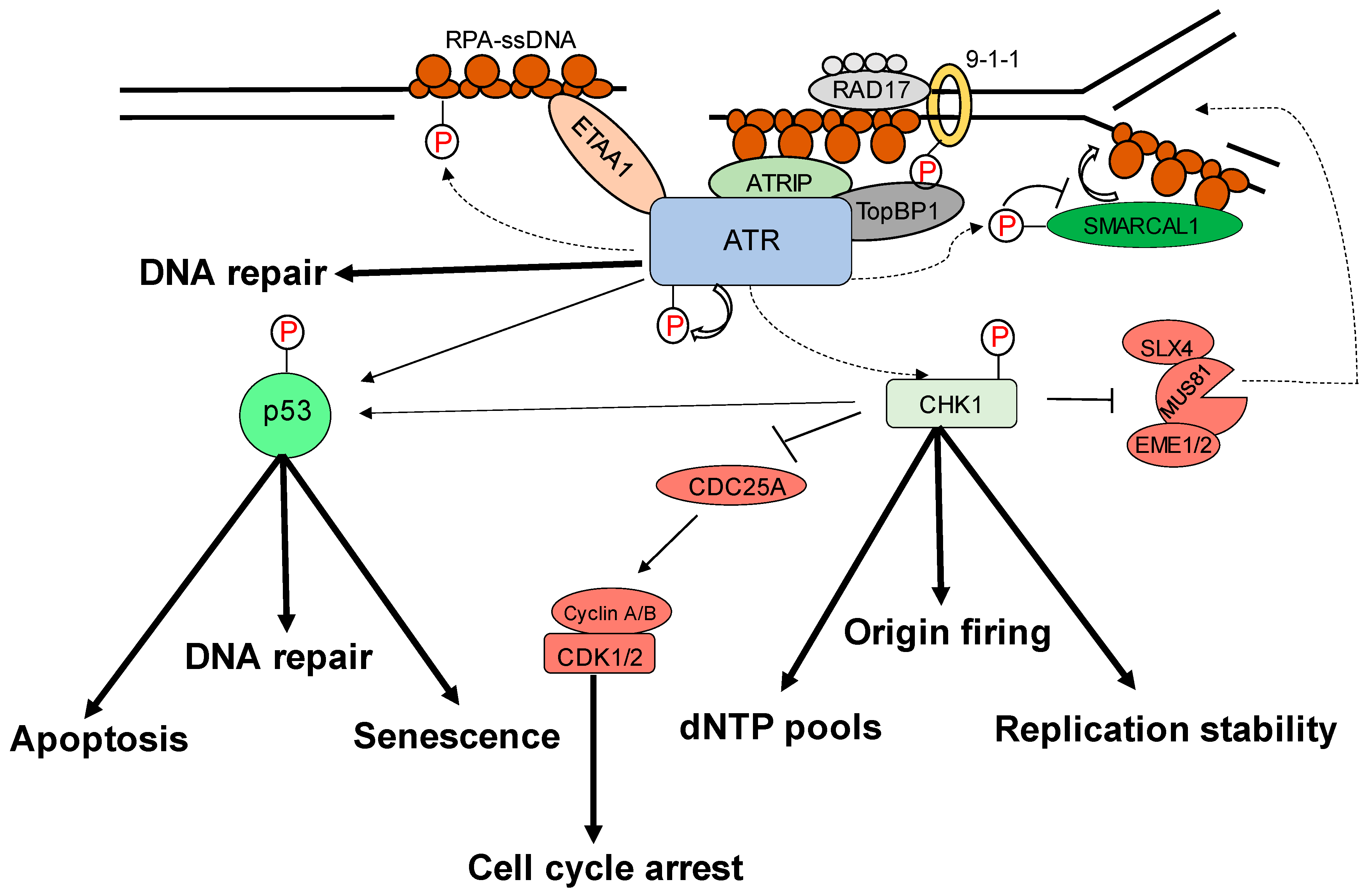
2. Role of ATR–Chk1 Pathway in Oncogenesis
3. Cytoplasmic Functions of ATR
4. ATR Regulation of Nucleus Mechanics
5. ATR Regulation of Autophagy
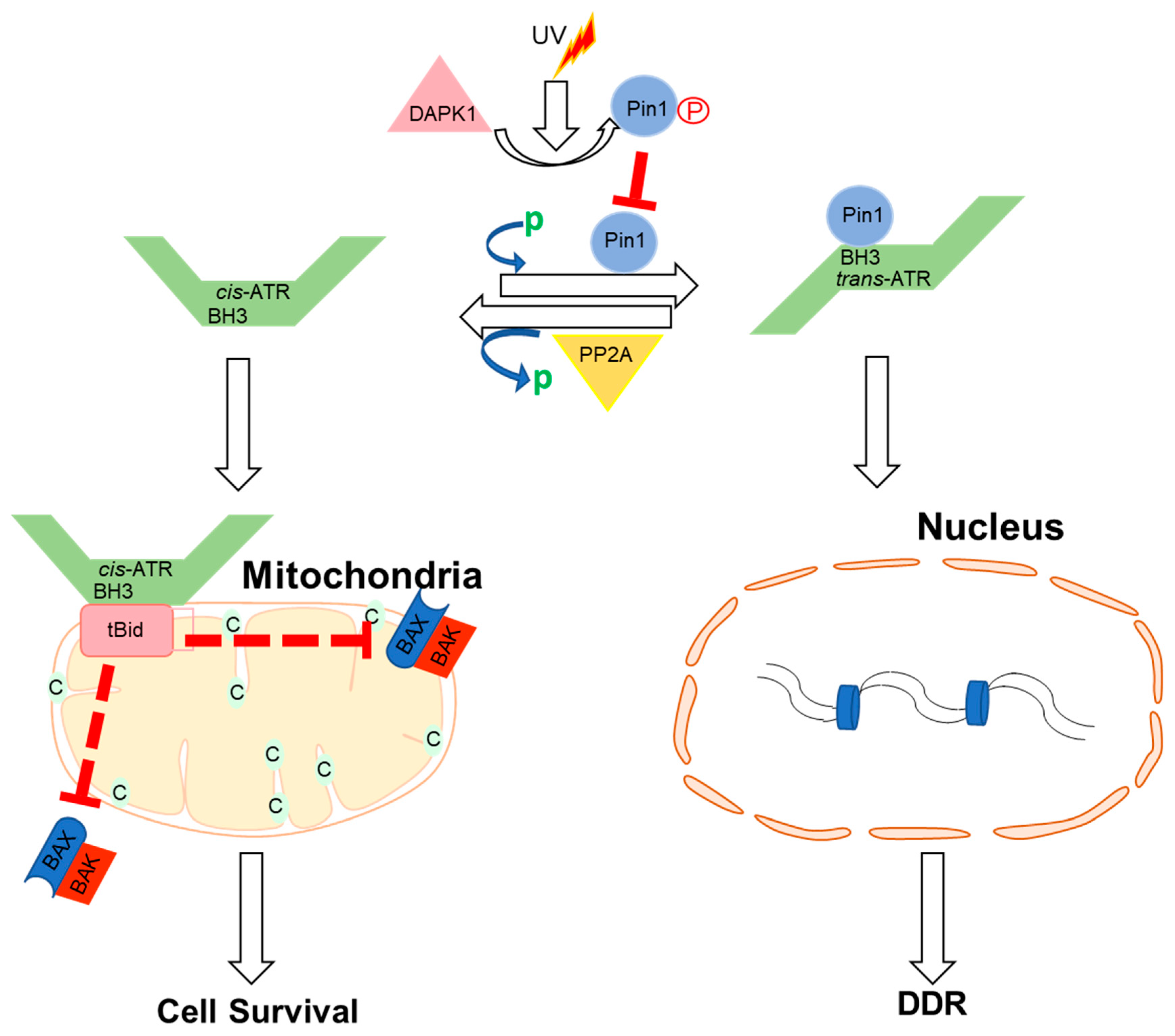
6. The cis-ATR Anti-Apoptotic Role Drives Oncogenesis in Dividing Cells
7. ATR: A Therapeutic Target
7.1. Chemotherapeutic Effect by ATR–CHK1 Inhibition
7.2. ATR Kinase Inhibitors
7.3. ATR Kinase Inhibitors in Clinical Trials
8. ATR Isomerization Potential Therapeutic Target
9. Prospects
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shechter, D.; Costanzo, V.; Gautier, J. Regulation of DNA replication by ATR: Signaling in response to DNA intermediates. DNA Repair. 2004, 3, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Biswas, H.; Goto, G.; Wang, W.; Sung, P.; Sugimoto, K. Ddc2ATRIP promotes Mec1ATR activation at RPA-ssDNA tracts. PLoS Genet. 2019, 15, e1008294. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in Checkpoint Signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Burrows, A.E.; Elledge, S.J. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes. Dev. 2008, 22, 1416–1421. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell. Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Wu, X.; Shell, S.M.; Zou, Y. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene 2005, 24, 4728–4735. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 Activates the ATR-ATRIP Complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Laura, A.; Boltz, L.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes. Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Mordes, D.A.; Cortez, D. Activation of ATR and related PIKKs. Cell. Cycle 2008, 7, 2809–2812. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef]
- Ma, K.; Wu, H.; Li, P.; Li, B. LC3-II may mediate ATR-induced mitophagy in dopaminergic neurons through SQSTM1/p62 pathway. Acta Biochim. Biophys. Sin. 2018, 50, 1047–1061. [Google Scholar] [CrossRef]
- Lam, M.H.; Rosen, J.M. Chk1 versus Cdc25: Chking one’s levels of cellular proliferation. Cell Cycle 2004, 3, 1355–1357. [Google Scholar] [CrossRef]
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169. [Google Scholar] [CrossRef]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes. Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., 3rd; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.; Powell, S.N.; Iliakis, G.; Wang, Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004, 64, 7139–7143. [Google Scholar] [CrossRef]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes. Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shell, S.M.; Liu, Y.; Zou, Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene 2007, 26, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shell, S.M.; Yang, Z.; Zou, Y. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res. 2006, 66, 2997–3005. [Google Scholar] [CrossRef]
- Shell, S.M.; Li, Z.; Shkriabai, N.; Kvaratskhelia, M.; Brosey, C.; Serrano, M.A.; Chazin, W.J.; Musich, P.R.; Zou, Y. Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with xeroderma pigmentosum group A. J. Biol. Chem. 2009, 284, 24213–24222. [Google Scholar] [CrossRef]
- Lecona, E.; Fernández-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes. Dev. 2000, 14, 397–402. [Google Scholar] [CrossRef]
- Menolfi, D.; Jiang, W.; Lee, B.J.; Moiseeva, T.; Shao, Z.; Estes, V.; Frattini, M.G.; Bakkenist, C.J.; Zha, S. Kinase-dead ATR differs from ATR loss by limiting the dynamic exchange of ATR and RPA. Nat. Commun. 2018, 9, 5351. [Google Scholar] [CrossRef]
- Ruzankina, Y.; Pinzon-Guzman, C.; Asare, A.; Ong, T.; Pontano, L.; Cotsarelis, G.; Zediak, V.P.; Velez, M.; Bhandoola, A.; Brown, E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 2007, 1, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes. Dev. 2003, 17, 615–628. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef]
- Höglund, A.; Nilsson, L.M.; Muralidharan, S.V.; Hasvold, L.A.; Merta, P.; Rudelius, M.; Nikolova, V.; Keller, U.; Nilsson, J.A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7067–7079. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef]
- Ferrao, P.T.; Bukczynska, E.P.; Johnstone, R.W.; McArthur, G.A. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 2012, 31, 1661–1672. [Google Scholar] [CrossRef]
- Walton, M.I.; Eve, P.D.; Hayes, A.; Valenti, M.R.; De Haven Brandon, A.K.; Box, G.; Hallsworth, A.; Smith, E.L.; Boxall, K.J.; Lainchbury, M.; et al. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5650–5661. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakti, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS ONE 2013, 8, e57098. [Google Scholar] [CrossRef]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef]
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A Synthetic Lethal Screen Identifies DNA Repair Pathways that Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS ONE 2015, 10, e0125482. [Google Scholar] [CrossRef]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Hammond, E.M.; Dorie, M.J.; Giaccia, A.J. Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Res. 2004, 64, 6556–6562. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar] [CrossRef]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. 2012, 13, 1072–1081. [Google Scholar] [CrossRef]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef]
- López-Contreras, A.J.; Fernandez-Capetillo, O. The ATR barrier to replication-born DNA damage. DNA Repair. 2010, 9, 1249–1255. [Google Scholar] [CrossRef]
- Ruzankina, Y.; Schoppy, D.W.; Asare, A.; Clark, C.E.; Vonderheide, R.H.; Brown, E.J. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat. Genet. 2009, 41, 1144–1149. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.G.; Muschel, R.J.; Gupta, A.K.; Hahn, S.M.; Bernhard, E.J. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 2003, 22, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Makinwa, Y.; Cartwright, B.M.; Musich, P.R.; Li, Z.; Biswas, H.; Zou, Y. PP2A Regulates Phosphorylation-Dependent Isomerization of Cytoplasmic and Mitochondrial-Associated ATR by Pin1 in DNA Damage Responses. Front. Cell Dev. Biol. 2020, 8, 813. [Google Scholar] [CrossRef]
- Murga, M.; Bunting, S.; Montaña, M.F.; Soria, R.; Mulero, F.; Cañamero, M.; Lee, Y.; McKinnon, P.J.; Nussenzweig, A.; Fernandez-Capetillo, O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009, 41, 891–898. [Google Scholar] [CrossRef]
- Biswas, H.; Zhao, S.J.; Makinwa, Y.; Bassett, J.S.; Musich, P.R.; Liu, J.Y.; Zou, Y. Prolyl Isomerization-Mediated Conformational Changes Define ATR Subcellular Compartment-Specific Functions. Front. Cell. Dev. Biol. 2022, 10, 826576. [Google Scholar] [CrossRef] [PubMed]
- Musich, P.R.; Li, Z.; Zou, Y. Xeroderma Pigmentosa Group A (XPA), Nucleotide Excision Repair and Regulation by ATR in Response to Ultraviolet Irradiation. Adv. Exp. Med. Biol. 2017, 996, 41–54. [Google Scholar] [CrossRef]
- Makinwa, Y.; Musich, P.R.; Zou, Y. Phosphorylation-Dependent Pin1 Isomerization of ATR: Its Role in Regulating ATR’s Anti-apoptotic Function at Mitochondria, and the Implications in Cancer. Front. Cell. Dev. Biol. 2020, 8, 281. [Google Scholar] [CrossRef]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315. [Google Scholar] [CrossRef]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- Kidiyoor, G.R.; Li, Q.; Bastianello, G.; Bruhn, C.; Giovannetti, I.; Mohamood, A.; Beznoussenko, G.V.; Mironov, A.; Raab, M.; Piel, M.; et al. ATR is essential for preservation of cell mechanics and nuclear integrity during interstitial migration. Nat. Commun. 2020, 11, 4828. [Google Scholar] [CrossRef]
- Tan, X.; Zhou, F.; Wan, J.; Hang, J.; Chen, Z.; Li, B.; Zhang, C.; Shao, K.; Jiang, P.; Shi, S.; et al. Pin1 expression contributes to lung cancer: Prognosis and carcinogenesis. Cancer Biol. 2010, 9, 111–119. [Google Scholar] [CrossRef]
- Liu, M.; Zeng, T.; Zhang, X.; Liu, C.; Wu, Z.; Yao, L.; Xie, C.; Xia, H.; Lin, Q.; Xie, L.; et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 2018, 9, 4139. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Ayala, G.; Wang, D.; Wulf, G.; Frolov, A.; Li, R.; Sowadski, J.; Wheeler, T.M.; Lu, K.P.; Bao, L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003, 63, 6244–6251. [Google Scholar]
- He, J.; Zhou, F.; Shao, K.; Hang, J.; Wang, H.; Rayburn, E.; Xiao, Z.X.; Lee, S.W.; Xue, Q.; Feng, X.L.; et al. Overexpression of Pin1 in non-small cell lung cancer (NSCLC) and its correlation with lymph node metastases. Lung Cancer 2007, 56, 51–58. [Google Scholar] [CrossRef]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Luo, M.L.; Gong, C.; Chen, C.H.; Lee, D.Y.; Hu, H.; Huang, P.; Yao, Y.; Guo, W.; Reinhardt, F.; Wulf, G.; et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 2014, 74, 3603–3616. [Google Scholar] [CrossRef]
- Rustighi, A.; Zannini, A.; Tiberi, L.; Sommaggio, R.; Piazza, S.; Sorrentino, G.; Nuzzo, S.; Tuscano, A.; Eterno, V.; Benvenuti, F.; et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol. Med. 2014, 6, 99–119. [Google Scholar] [CrossRef]
- Xu, M.; Cheung, C.C.; Chow, C.; Lun, S.W.; Cheung, S.T.; Lo, K.W. Overexpression of PIN1 Enhances Cancer Growth and Aggressiveness with Cyclin D1 Induction in EBV-Associated Nasopharyngeal Carcinoma. PLoS ONE 2016, 11, e0156833. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Yamamotoya, T.; Ueda, K.; Ono, H.; Inoue, M.K.; Matsunaga, Y.; Kushiyama, A.; Sakoda, H.; Fujishiro, M.; Matsubara, A.; et al. Prolyl isomerase Pin1 in metabolic reprogramming of cancer cells. Cancer Lett. 2020, 470, 106–114. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.R.; Yang, H.Y.; Li, X.Z.; Jie, M.M.; Hu, C.J.; Wu, Y.Y.; Yang, S.M.; Yang, Y.B. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef]
- Hennig, L.; Christner, C.; Kipping, M.; Schelbert, B.; Rucknagel, K.P.; Grabley, S.; Kullertz, G.; Fischer, G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 1998, 37, 5953–5960. [Google Scholar] [CrossRef]
- Campaner, E.; Rustighi, A.; Zannini, A.; Cristiani, A.; Piazza, S.; Ciani, Y.; Kalid, O.; Golan, G.; Baloglu, E.; Shacham, S.; et al. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 2017, 8, 15772. [Google Scholar] [CrossRef]
- Estey, E.; Thall, P.F.; Pierce, S.; Kantarjian, H.; Keating, M. Treatment of newly diagnosed acute promyelocytic leukemia without cytarabine. J. Clin. Oncol. 1997, 15, 483–490. [Google Scholar] [CrossRef]
- Budd, G.T.; Adamson, P.C.; Gupta, M.; Homayoun, P.; Sandstrom, S.K.; Murphy, R.F.; McLain, D.; Tuason, L.; Peereboom, D.; Bukowski, R.M.; et al. Phase I/II trial of all-trans retinoic acid and tamoxifen in patients with advanced breast cancer. Clin. Cancer Res. 1998, 4, 635–642. [Google Scholar]
- Shen, Z.X.; Shi, Z.Z.; Fang, J.; Gu, B.W.; Li, J.M.; Zhu, Y.M.; Shi, J.Y.; Zheng, P.Z.; Yan, H.; Liu, Y.F.; et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 5328–5335. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014, 24, 1033–1049. [Google Scholar] [CrossRef]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef]
- Lian, X.; Lin, Y.M.; Kozono, S.; Herbert, M.K.; Li, X.; Yuan, X.; Guo, J.; Guo, Y.; Tang, M.; Lin, J.; et al. Pin1 inhibition exerts potent activity against acute myeloid leukemia through blocking multiple cancer-driving pathways. J. Hematol. Oncol. 2018, 11, 73. [Google Scholar] [CrossRef]
- Muindi, J.; Frankel, S.R.; Miller, W.H., Jr.; Jakubowski, A.; Scheinberg, D.A.; Young, C.W.; Dmitrovsky, E.; Warrell, R.P., Jr. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: Implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood 1992, 79, 299–303. [Google Scholar] [CrossRef]
- Decensi, A.; Robertson, C.; Guerrieri-Gonzaga, A.; Serrano, D.; Cazzaniga, M.; Mora, S.; Gulisano, M.; Johansson, H.; Galimberti, V.; Cassano, E.; et al. Randomized double-blind 2 × 2 trial of low-dose tamoxifen and fenretinide for breast cancer prevention in high-risk premenopausal women. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3749–3756. [Google Scholar] [CrossRef]
- Arrieta, O.; Gonzalez-De la Rosa, C.H.; Arechaga-Ocampo, E.; Villanueva-Rodriguez, G.; Ceron-Lizarraga, T.L.; Martinez-Barrera, L.; Vazquez-Manriquez, M.E.; Rios-Trejo, M.A.; Alvarez-Avitia, M.A.; Hernandez-Pedro, N.; et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3463–3471. [Google Scholar] [CrossRef]
- Moore, J.D.; Potter, A. Pin1 inhibitors: Pitfalls, progress and cellular pharmacology. Bioorg. Med. Chem. Lett. 2013, 23, 4283–4291. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Estey, E.; Pierce, S.; Cortes, J.; Lopez-Berestein, G.; Ravandi, F. Single-agent liposomal all-trans-retinoic Acid as initial therapy for acute promyelocytic leukemia: 13-year follow-up data. Clin. Lymphoma Myeloma Leuk. 2014, 14, e47–e49. [Google Scholar] [CrossRef]
- Khoei, S.G.; Mohammadi, C.; Mohammadi, Y.; Sameri, S.; Najafi, R. Prognostic value of peptidyl-prolyl cis-trans isomerase 1 (PIN1) in human malignant tumors. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2020, 22, 1067–1077. [Google Scholar] [CrossRef]
- Wulf, G.M.; Liou, Y.C.; Ryo, A.; Lee, S.W.; Lu, K.P. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 2002, 277, 47976–47979. [Google Scholar] [CrossRef]
- Zacchi, P.; Gostissa, M.; Uchida, T.; Salvagno, C.; Avolio, F.; Volinia, S.; Ronai, Z.; Blandino, G.; Schneider, C.; Del Sal, G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002, 419, 853–857. [Google Scholar] [CrossRef]
- Zheng, H.; You, H.; Zhou, X.Z.; Murray, S.A.; Uchida, T.; Wulf, G.; Gu, L.; Tang, X.; Lu, K.P.; Xiao, Z.X. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002, 419, 849–853. [Google Scholar] [CrossRef]
- Mantovani, F.; Zannini, A.; Rustighi, A.; Del Sal, G. Interaction of p53 with prolyl isomerases: Healthy and unhealthy relationships. Biochim. Biophys. Acta 2015, 1850, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Choi, B.Y.; Surh, Y.J. Dual Roles of Pin1 in Cancer Development and Progression. Curr. Pharm. Des. 2017, 23, 4422–4425. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Vance, S.; Liu, E.; Zhao, L.; Parsels, J.D.; Parsels, L.A.; Brown, J.L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 2011, 10, 4321–4329. [Google Scholar] [CrossRef]
- Dent, P.; Tang, Y.; Yacoub, A.; Dai, Y.; Fisher, P.B.; Grant, S. CHK1 inhibitors in combination chemotherapy: Thinking beyond the cell cycle. Mol. Interv. 2011, 11, 133–140. [Google Scholar] [CrossRef]
- Bunch, R.T.; Eastman, A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 791–797. [Google Scholar]
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110. [Google Scholar] [CrossRef]
- Sha, S.K.; Sato, T.; Kobayashi, H.; Ishigaki, M.; Yamamoto, S.; Sato, H.; Takada, A.; Nakajyo, S.; Mochizuki, Y.; Friedman, J.M.; et al. Cell cycle phenotype-based optimization of G2-abrogating peptides yields CBP501 with a unique mechanism of action at the G2 checkpoint. Mol. Cancer Ther. 2007, 6, 147–153. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Petsalaki, E.; Akoumianaki, T.; Black, E.J.; Gillespie, D.A.; Zachos, G. Phosphorylation at serine 331 is required for Aurora B activation. J. Cell Biol. 2011, 195, 449–466. [Google Scholar] [CrossRef]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef]
- Neumann, J.; Boerries, M.; Köhler, R.; Giaisi, M.; Krammer, P.H.; Busch, H.; Li-Weber, M. The natural anticancer compound rocaglamide selectively inhibits the G1-S-phase transition in cancer cells through the ATM/ATR-mediated Chk1/2 cell cycle checkpoints. Int. J. Cancer 2014, 134, 1991–2002. [Google Scholar] [CrossRef]
- Shen, T.; Huang, S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anti-Cancer Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, F.; Zhang, W.; Chen, L.; Gao, N.; Men, Y.; Xu, X.; Jiang, Y. Harmine suppresses homologous recombination repair and inhibits proliferation of hepatoma cells. Cancer Biol. Ther. 2015, 16, 1585–1592. [Google Scholar] [CrossRef]
- Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N.J. Targeting the ATR-CHK1 axis in cancer therapy. Cancers 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef]
- Wu, L.Y.; Lu, H.F.; Chou, Y.C.; Shih, Y.L.; Bau, D.T.; Chen, J.C.; Hsu, S.C.; Chung, J.G. Kaempferol induces DNA damage and inhibits DNA repair associated protein expressions in human promyelocytic leukemia HL-60 cells. Am. J. Chin. Med. 2015, 43, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Wu, X.; Liu, Y.; Liu, Y.; Zhou, J.; Jiang, X.; Zhang, L.; He, X.; Ma, L. Curcumin-Induced DNA Demethylation in Human Gastric Cancer Cells Is Mediated by the DNA-Damage Response Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 2543504. [Google Scholar] [CrossRef]
- Ogiwara, H.; Ui, A.; Shiotani, B.; Zou, L.; Yasui, A.; Kohno, T. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis 2013, 34, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Singh, R.P.; Agarwal, C.; Siriwardana, S.; Sclafani, R.A.; Agarwal, R. Resveratrol causes Cdc2-tyr15 phosphorylation via ATM/ATR–Chk1/2–Cdc25C pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells. Carcinogenesis 2005, 26, 1978–1987. [Google Scholar] [CrossRef]
- Leon-Galicia, I.; Diaz-Chavez, J.; Garcia-Villa, E.; Uribe-Figueroa, L.; Hidalgo-Miranda, A.; Herrera, L.A.; Alvarez-Rios, E.; Garcia-Mena, J.; Gariglio, P. Resveratrol induces downregulation of DNA repair genes in MCF-7 human breast cancer cells. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. (ECP) 2013, 22, 11–20. [Google Scholar] [CrossRef]
- George, V.C.; Rupasinghe, H.P.V. Apple Flavonoids Suppress Carcinogen-Induced DNA Damage in Normal Human Bronchial Epithelial Cells. Oxidative Med. Cell. Longev. 2017, 2017, 1767198. [Google Scholar] [CrossRef] [PubMed]
- Chueh, F.S.; Chen, Y.L.; Hsu, S.C.; Yang, J.S.; Hsueh, S.C.; Ji, B.C.; Lu, H.F.; Chung, J.G. Triptolide induced DNA damage in A375.S2 human malignant melanoma cells is mediated via reduction of DNA repair genes. Oncol. Rep. 2013, 29, 613–618. [Google Scholar] [CrossRef]
- Peng, Z.G.; Yao, Y.B.; Yang, J.; Tang, Y.L.; Huang, X. Mangiferin induces cell cycle arrest at G2/M phase through ATR-Chk1 pathway in HL-60 leukemia cells. Genet. Mol. Res. GMR 2015, 14, 4989–5002. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Lee, A.Y.; Chou, W.C.; Wu, C.C.; Tseng, C.N.; Liu, K.Y.; Lin, W.L.; Chang, F.R.; Chuang, D.W.; Hunyadi, A.; et al. Inhibition of ATR-dependent signaling by protoapigenone and its derivative sensitizes cancer cells to interstrand cross-link-generating agents in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1443–1453. [Google Scholar] [CrossRef]
- Blasina, A.; Price, B.D.; Turenne, G.A.; McGowan, C.H. Caffeine inhibits the checkpoint kinase ATM. Curr. Biol. CB 1999, 9, 1135–1138. [Google Scholar] [CrossRef]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell. Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef]
- Vávrová, J.; Zárybnická, L.; Lukášová, E.; Řezáčová, M.; Novotná, E.; Sinkorová, Z.; Tichý, A.; Pejchal, J.; Durišová, K. Inhibition of ATR kinase with the selective inhibitor VE-821 results in radiosensitization of cells of promyelocytic leukaemia (HL-60). Radiat. Environ. Biophys. 2013, 52, 471–479. [Google Scholar] [CrossRef]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014, 74, 6968–6979. [Google Scholar] [CrossRef]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.D.; Blades, K.; Foote, K.M.; Guichard, S.M.; Jewsbury, P.J.; McGuire, T.; Nissink, J.W.; Odedra, R.; Tam, K.; Thommes, P. Discovery of AZD6738, a potent and selective inhibitor with the potential to test the clinical efficacy of ATR kinase inhibition in cancer patients. Cancer Res. 2013, 73, 2348. [Google Scholar] [CrossRef]
- Clack, G.; Lau, A.; Pierce, A.; Smith, S.; Stephens, C. ATR inhibitor AZD6738. Ann. Oncol. 2015, 26, ii8. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Petermann, E.; Yates, E.; Brown, J.; Lau, A.; Stankovic, T. Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet 2015, 385, S58. [Google Scholar] [CrossRef]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A synthetic lethal screen reveals enhanced sensitivity to ATR inhibitor treatment in mantle cell lymphoma with ATM loss-of-function. Mol. Cancer Res. MCR 2015, 13, 120–129. [Google Scholar] [CrossRef]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef]
- Konstantinidou, G.; Bey, E.A.; Rabellino, A.; Schuster, K.; Maira, M.S.; Gazdar, A.F.; Amici, A.; Boothman, D.A.; Scaglioni, P.P. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009, 69, 7644–7652. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Li, Z.; Li, H.; Zhang, J.T. Critical residue that promotes protein dimerization: A story of partially exposed Phe25 in 14-3-3σ. J. Chem. Inf. Model. 2011, 51, 2612–2625. [Google Scholar] [CrossRef]
- Sangster-Guity, N.; Conrad, B.H.; Papadopoulos, N.; Bunz, F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011, 30, 2526–2533. [Google Scholar] [CrossRef]
- Wouters, B.G.; Brown, J.M. Cells at Intermediate Oxygen Levels Can Be More Important Than the “Hypoxic Fraction” in Determining Tumor Response to Fractionated Radiotherapy. Radiat. Res. 1997, 147, 541–550. [Google Scholar] [CrossRef]
- Sprong, D.; Janssen, H.L.; Vens, C.; Begg, A.C. Resistance of hypoxic cells to ionizing radiation is influenced by homologous recombination status. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Jacq, X.; Smith, L.; Brown, E.; Hughes, A.; Odedra, R.; Heathcote, D.; Barnes, J.; Powell, S.; Maguire, S.; Pearson, V.; et al. Abstract 1823: AZ20, a novel potent and selective inhibitor of ATR kinase with in vivo antitumour activity. Cancer Res. 2012, 72, 1823. [Google Scholar] [CrossRef]
- Guichard, S.M.; Brown, E.; Odedra, R.; Hughes, A.; Heathcote, D.; Barnes, J.; Lau, A.; Powell, S.; Jones, C.D.; Nissink, W.; et al. Abstract 3343: The pre-clinical in vitro and in vivo activity of AZD6738: A potent and selective inhibitor of ATR kinase. Cancer Res. 2013, 73, 3343. [Google Scholar] [CrossRef]
- Ibarra, M.S.; Borini Etichetti, C.; Di Benedetto, C.; Rosano, G.L.; Margarit, E.; Del Sal, G.; Mione, M.; Girardini, J. Dynamic regulation of Pin1 expression and function during zebrafish development. PLoS ONE 2017, 12, e0175939. [Google Scholar] [CrossRef]
- Lee, B.; Lee, H.J.; Cho, H.Y.; Suh, D.H.; Kim, K.; No, J.H.; Kim, H.; Kim, Y.B. Ataxia-Telangiectasia and RAD3-Related and Ataxia-Telangiectasia-Mutated Proteins in Epithelial Ovarian Carcinoma: Their Expression and Clinical Significance. Anticancer Res. 2015, 35, 3909–3916. [Google Scholar]
- Freeman, A.K.; Monteiro, A.N.A. Phosphatases in the cellular response to DNA damage. Cell. Commun. Signal. 2010, 8, 27. [Google Scholar] [CrossRef]
- Harris, D.R.; Bunz, F. Protein phosphatases and the dynamics of the DNA damage response. Cell. Cycle 2010, 9, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Palii, S.S.; Cui, Y.; Innes, C.L.; Paules, R.S. Dissecting cellular responses to irradiation via targeted disruptions of the ATM-CHK1-PP2A circuit. Cell. Cycle 2013, 12, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Bruhn, C.; Peretti, M.; Cassani, C.; Carotenuto, W.V.; Elgendy, M.; Shubassi, G.; Lucca, C.; Bermejo, R.; Varasi, M. PP2A controls genome integrity by integrating nutrient-sensing and metabolic pathways with the DNA damage response. Mol. Cell 2017, 67, 266–281.e264. [Google Scholar] [CrossRef] [PubMed]
- Merigliano, C.; Marzio, A.; Renda, F.; Somma, M.P.; Gatti, M.; Vernì, F. A role for the twins protein phosphatase (PP2A-B55) in the maintenance of Drosophila genome integrity. Genetics 2017, 205, 1151–1167. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.; Villoria, M.T.; Alonso-Rodríguez, E.; Clemente-Blanco, A. Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response. Cell. Stress. 2019, 3, 70. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jonnalagadda, J.C.; Douglas, P.; Young, D.; Ye, R.; Moorhead, G.B.; Lees-Miller, S.P.; Khanna, K.K. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004, 23, 4451–4461. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, I.; Smirnova, A.; Krutilina, R.; Svetlova, M.; Solovjeva, L.; Nikiforov, A.; Oei, S.; Zalenskaya, I.; Yau, P.; Bradbury, E. Dephosphorylation of histone γ-H2AX during repair of DNA double-strand breaks in mammalian cells and its inhibition by calyculin A. Radiat. Res. 2003, 160, 309–317. [Google Scholar] [CrossRef]
- Chowdhury, D.; Keogh, M.-C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. γ-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef]
- Keogh, M.-C.; Kim, J.-A.; Downey, M.; Fillingham, J.; Chowdhury, D.; Harrison, J.C.; Onishi, M.; Datta, N.; Galicia, S.; Emili, A. A phosphatase complex that dephosphorylates γH2AX regulates DNA damage checkpoint recovery. Nature 2006, 439, 497–501. [Google Scholar] [CrossRef]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef]
- Andrabi, S.; Gjoerup, O.V.; Kean, J.A.; Roberts, T.M.; Schaffhausen, B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. USA 2007, 104, 19011–19016. [Google Scholar] [CrossRef] [PubMed]
- Guenebeaud, C.; Goldschneider, D.; Castets, M.; Guix, C.; Chazot, G.; Delloye-Bourgeois, C.; Eisenberg-Lerner, A.; Shohat, G.; Zhang, M.; Laudet, V. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol. Cell 2010, 40, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Luo, W.; Zeng, C.; Zhang, Y.; Wang, L.; Yao, W.; Nie, C. PP2A mediates apoptosis or autophagic cell death in multiple myeloma cell lines. Oncotarget 2017, 8, 80770. [Google Scholar] [CrossRef]
- Janssens, V.; Rebollo, A. The role and therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic signalling networks in human cancer cells. Curr. Mol. Med. 2012, 12, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Parsels, L.A.; Karnak, D.; Davis, M.A.; Parsels, J.D.; Marsh, A.C.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Sun, Y.; et al. Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.K.; Lu, J.; Graves, C.A.; Huntoon, K.; Frerich, J.M.; Hanson, R.H.; Wang, X.; Hong, C.S.; Ho, W.; Feldman, M.J.; et al. Protein Phosphatase 2A Inhibition with LB100 Enhances Radiation-Induced Mitotic Catastrophe and Tumor Growth Delay in Glioblastoma. Mol. Cancer Ther. 2015, 14, 1540–1547. [Google Scholar] [CrossRef]
- Ho, W.S.; Sizdahkhani, S.; Hao, S.; Song, H.; Seldomridge, A.; Tandle, A.; Maric, D.; Kramp, T.; Lu, R.; Heiss, J.D.; et al. LB-100, a novel Protein Phosphatase 2A (PP2A) inhibitor, sensitizes malignant meningioma cells to the therapeutic effects of radiation. Cancer Lett. 2018, 415, 217–226. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Mode of Action | IC50 | Cell Type | References |
|---|---|---|---|---|
| Rocaglamide-A (Roc-A) | Roc-A induces the rapid degradation of Cdc25A by activation of the ATM/ATR–Chk1/Chk2 checkpoint pathway | 50–100 nM | Acute T cell leukemia lines Jurkat-16, CEM, Molt-4, and DND-41; the T lymphoma cell line Hut-78; the acute myeloid leukemia cell line HL-60; the Hodgkin lymphoma cell line L1236; the hepatocarcinoma cancer cell lines HepG2 and Huh7; the colorectal cancer cell lines HT-29 and HCT116; the prostate cancer cell line PC3; and the breast cancer cell line MCF-7 | [113,114] |
| Harmine | Harmine, a natural compound, negatively regulates HR but not NHEJ by interfering with Rad51 recruitment, resulting in severe cytotoxicity in hepatoma cells | 20 μM | Hep3B and HuH7 cell | [115] |
| Caffeine | Disrupted the G2-M and hastened the transition to mitosis, culminating in apoptosis in A549 cells | 1.1 mM | BCR/ABL leukemia cells | [116,117] |
| Schisandrin B (Sch-B) | inhibit ATR in A549 adenocarcinoma cells | 7.25 μM | A549 cells | [118] |
| Kaempferol | promoted DNA damage in HL-60 cells | 75 μM | HL-60 cells | [119] |
| Curcumin | Curcumin-induced DNA demethylation of human gastric cancer cells (hGCCs) was mediated by the damaged DNA repair-p53-p21/GADD45A-cyclin/CDK-Rb/E2F-DNMT1 axis | 20 μM | MGC-803 gastric cancer cells | [120] |
| Curcumin suppresses three DDR pathways by inhibiting histone acetyltransferases and ATR. Concordantly, curcumin sensitizes cancer cells to PARP inhibitors by enhancing apoptosis and mitotic catastrophe via inhibition of both the DNA damage checkpoint and DSB repair | 493 nM | HeLa (cervical cancer), U2OS (osteosarcoma), HT1080 (fibrosarcoma), HCT116 (colorectal cancer) cells and H1299 (non-small-cell lung carcinoma) | [121] | |
| Resveratrol | Resveratrol causes Cdc2-tyr15 phosphorylation via the ATM/ATR–Chk1/2–Cdc25C pathway as a central mechanism for S phase arrest | 50 μM | human ovarian carcinoma Ovcar-3 cells | [122] |
| Impedes the Rad51, BRCA1 (breast cancer 1), and BRCA2 expression involved in ATR–CHK1 activation | 150 μmol/L | MCF-7 cell | [123] | |
| Apple peel flavonoid fraction (AF4) | The apple peel flavonoid fraction (AF4) protects BEAS-2B cells against different carcinogens, including nicotine-derived nitrosamine ketones, via decreasing ATR–CHK1 signaling | 50 μg/mL | Normal human bronchial epithelial cells | [124] |
| Triptolide | Triptolide from Tripterygium wilfordii induces DNA damage in A375.S2 melanoma cells by ATR–CHK1 inhibition | 15–30 nM | The human malignant melanoma cell line (A375.S2) | [125] |
| Mangiferin | Mangiferin induces cell cycle arrest at the G2/M phase through the ATR–Chk1 pathway in HL-60 leukemia cells | 160 μM | HL-60 leukemia cells | [126] |
| Protoapigenone | The function of WYCs revealed that they have a potential ability to inhibit DDR, particularly on activation of Chk1 and Fanconi anemia group D2 protein (FANCD2), but not Chk2. In this way, WYCs inhibited ATR-mediated DNA damage checkpoint and repair | 8 μmol/L | MDA-MB-231 (breast adenocarcinoma) and A549 (lung adenocarcinoma | [127] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, H.; Makinwa, Y.; Zou, Y. Novel Cellular Functions of ATR for Therapeutic Targeting: Embryogenesis to Tumorigenesis. Int. J. Mol. Sci. 2023, 24, 11684. https://doi.org/10.3390/ijms241411684
Biswas H, Makinwa Y, Zou Y. Novel Cellular Functions of ATR for Therapeutic Targeting: Embryogenesis to Tumorigenesis. International Journal of Molecular Sciences. 2023; 24(14):11684. https://doi.org/10.3390/ijms241411684
Chicago/Turabian StyleBiswas, Himadri, Yetunde Makinwa, and Yue Zou. 2023. "Novel Cellular Functions of ATR for Therapeutic Targeting: Embryogenesis to Tumorigenesis" International Journal of Molecular Sciences 24, no. 14: 11684. https://doi.org/10.3390/ijms241411684
APA StyleBiswas, H., Makinwa, Y., & Zou, Y. (2023). Novel Cellular Functions of ATR for Therapeutic Targeting: Embryogenesis to Tumorigenesis. International Journal of Molecular Sciences, 24(14), 11684. https://doi.org/10.3390/ijms241411684