
Antioxidant Chimeric Molecules: Are Chemical Motifs Additive? The Case of a Selenium-Based Ligand

 , ,
, ,  ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
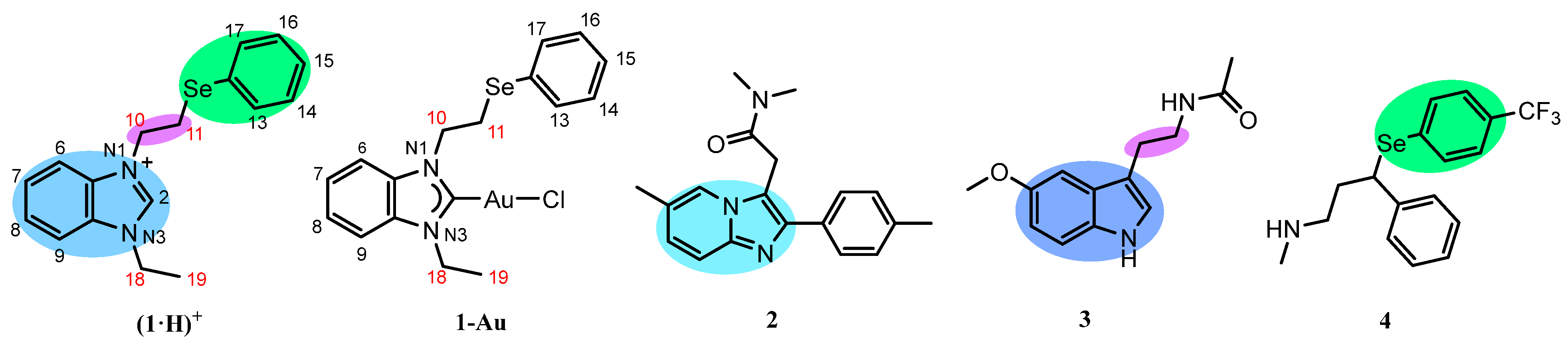
2.1. Rational Design of the Chimeric Compound
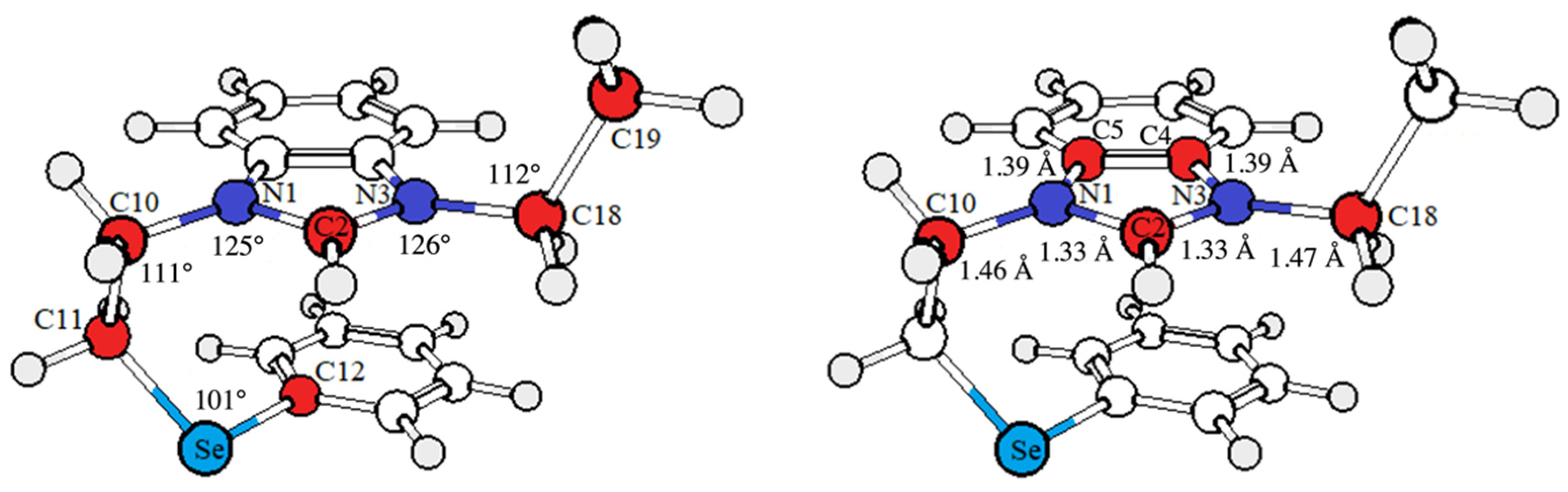
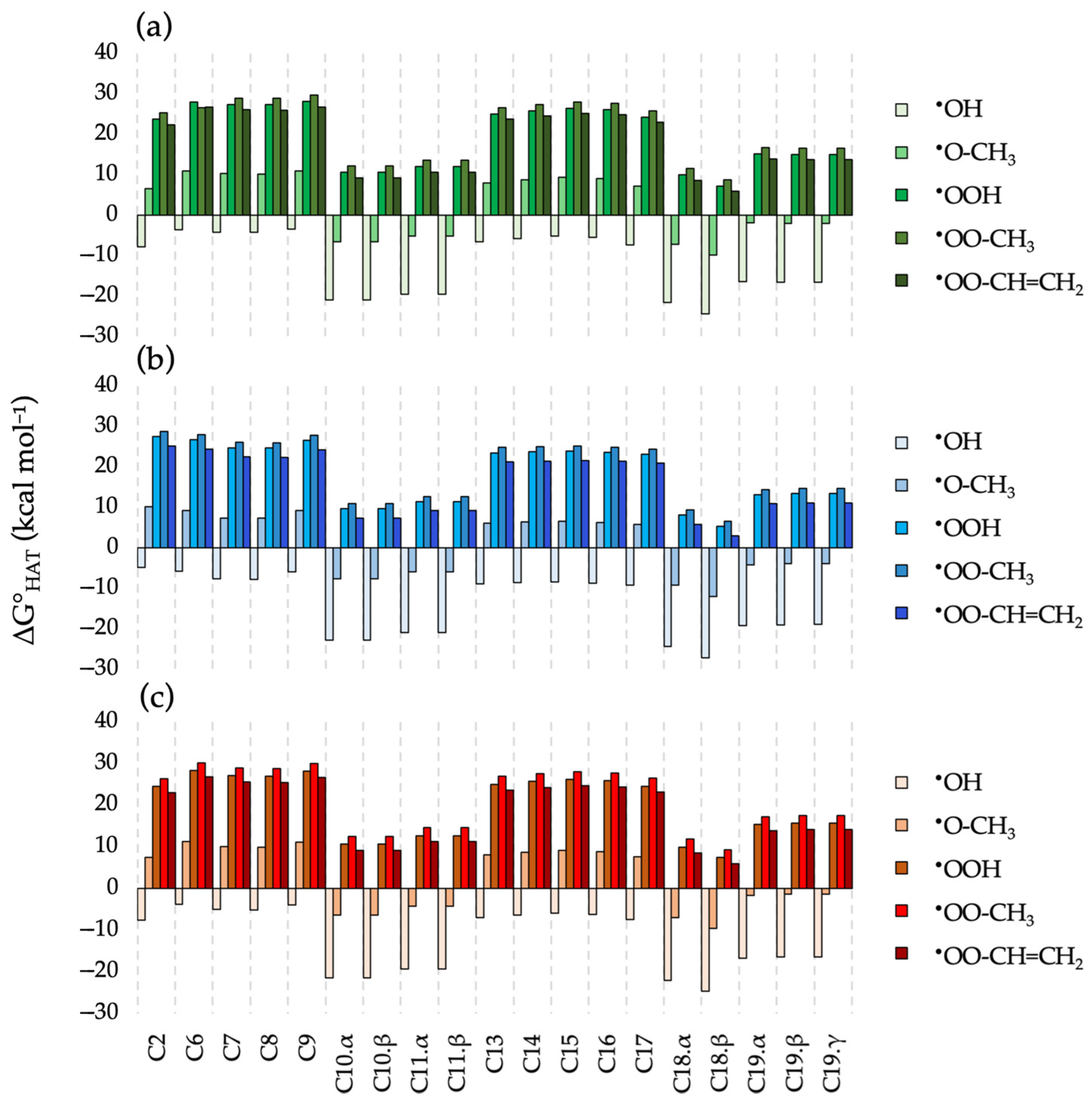
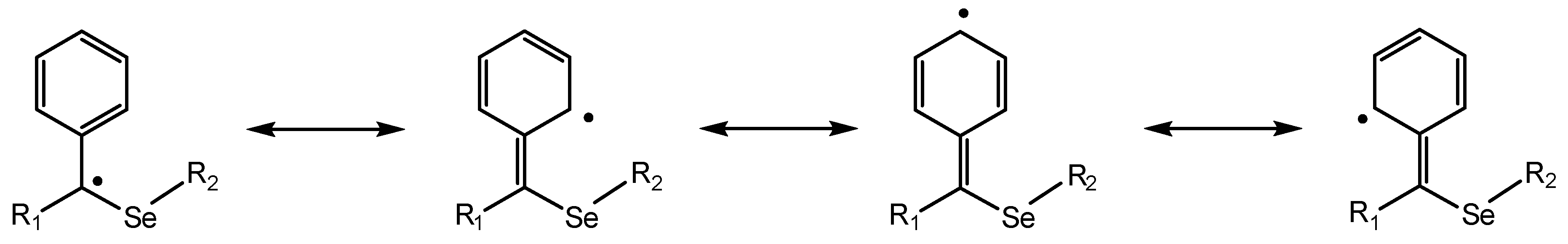
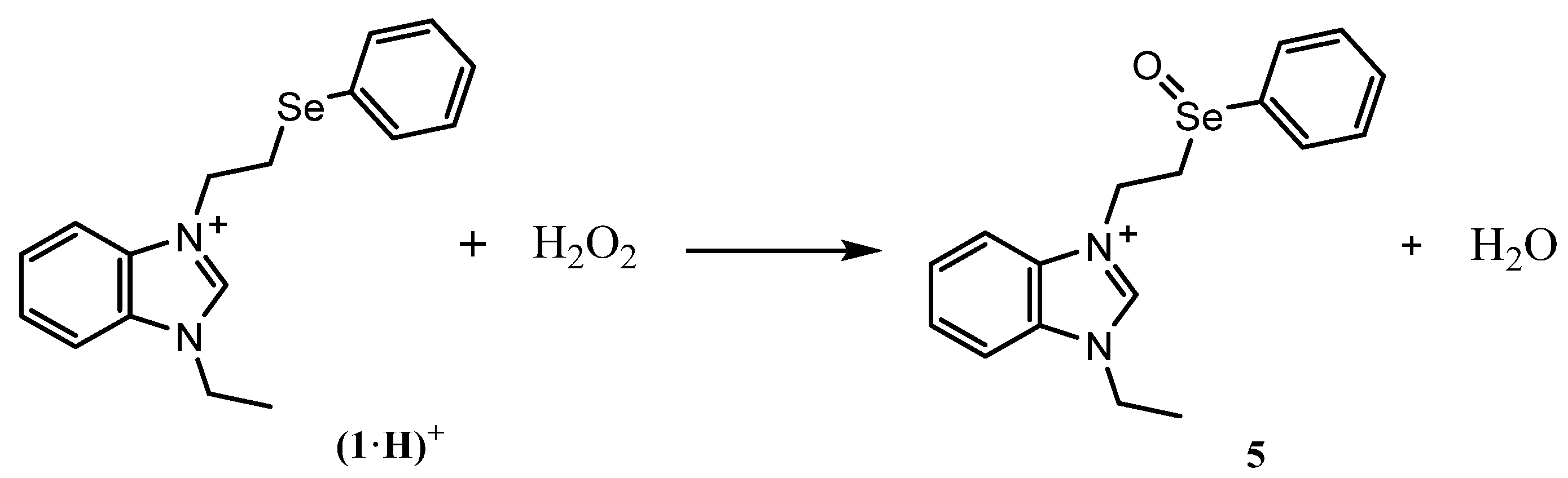
2.2. HAT Scavenging Mechanism of (1·H)+
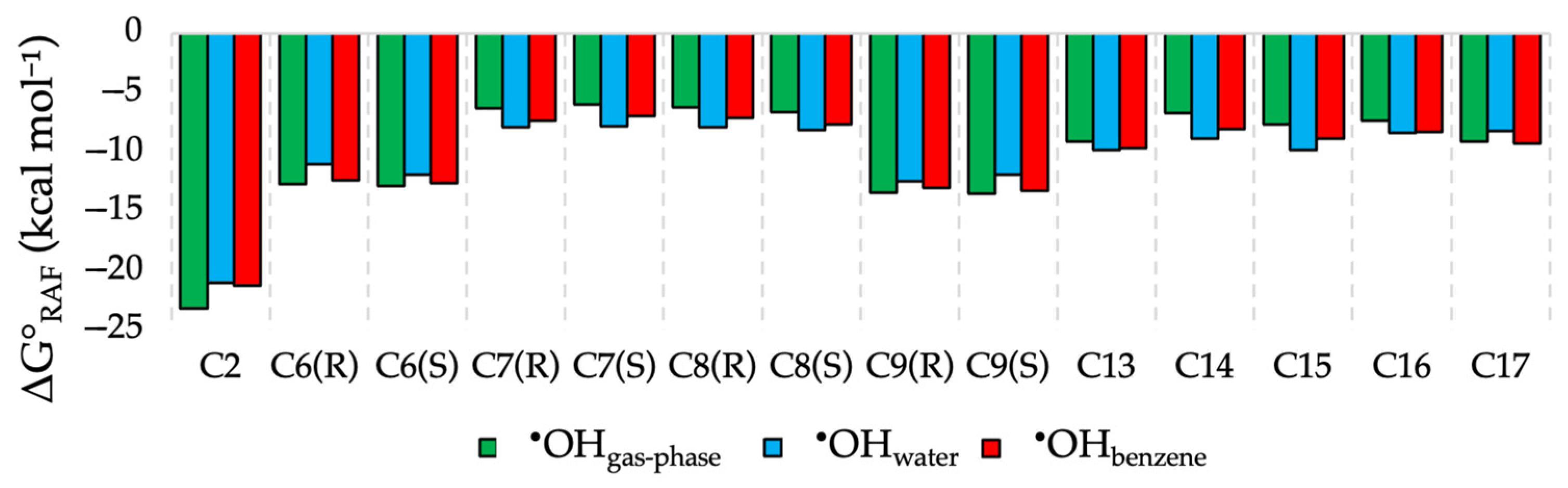
2.3. RAF Scavenging Mechanism of (1·H)+
2.4. HAT Scavenging Mechanism of 1-Au
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Lewis, D.F. Oxidative Stress: The Role of Cytochromes P450 in Oxygen Activation. J. Chem. Technol. Biotechnol. 2002, 77, 1095–1100. [Google Scholar] [CrossRef]
- Flohé, L. Glutathione Peroxidase: Fact and Fiction. Ciba Found. Symp. 1978, 65, 95–122. [Google Scholar]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative Diseases and Oxidative Stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2019, 24, 4771–4778. [Google Scholar] [CrossRef]
- Tobe, E. Mitochondrial Dysfunction, Oxidative Stress, and Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2013, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Emiliani, F.E.; Sedlak, T.W.; Sawa, A. Oxidative Stress and Schizophrenia. Curr. Opin. Psychiatry 2014, 27, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassen, M.; Youdim, M.B.H. Free Radical Scavengers: Chemical Concepts and Clinical Relevance. J. Neural. Transm. Suppl. 1999, 56, 193–210. [Google Scholar] [CrossRef]
- Eleutherio, E.C.A.; Silva Magalhães, R.S.; de Araújo Brasil, A.; Monteiro Neto, J.R.; de Holanda Paranhos, L. SOD1, More than Just an Antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Orian, L. The Glutathione Peroxidase Family: Discoveries and Mechanism. Free Radic. Biol. Med. 2022, 187, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Orian, L.; Flohé, L. Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology. Antioxidants 2021, 10, 1560. [Google Scholar] [CrossRef]
- Deisseroth, A.; Dounce, A.L. Catalase: Physical and Chemical Properties, Mechanism of Catalysis, and Physiological Role. Physiol. Rev. 1970, 50, 319–375. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food Antioxidants: Chemical Insights at the Molecular Level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a Natural Ally against Oxidative Stress: A Physicochemical Examination. J. Pineal. Res. 2011, 51, 1–16. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant Flavonoids: Classification, Distribution, Biosynthesis, and Antioxidant Activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef]
- Yoshida, Y.; Saito, Y.; Jones, L.S.; Shigeri, Y. Chemical Reactivities and Physical Effects in Comparison between Tocopherols and Tocotrienols: Physiological Significance and Prospects as Antioxidants. J. Biosci. Bioeng. 2007, 104, 439–445. [Google Scholar] [CrossRef]
- Cerezo, J.; Zúñiga, J.; Bastida, A.; Requena, A.; Cerón-Carrasco, J.P.; Eriksson, L.A. Antioxidant Properties of β-Carotene Isomers and Their Role in Photosystems: Insights from Ab Initio Simulations. J. Phys. Chem. A 2012, 116, 3498–3506. [Google Scholar] [CrossRef]
- Najafi, H.; Changizi-Ashtiyani, S.; Najafi, M. Antioxidant Activity of Omega-3 Derivatives and Their Delivery via Nanocages and Nanocones: DFT and Experimental in Vivo Investigation. J. Mol. Model. 2017, 23, 326. [Google Scholar] [CrossRef] [PubMed]
- Ahmadvand, H.; Mabuchi, H.; Nohara, A.; Kobayahi, J.; Kawashiri, M. Effects of Coenzyme Q(10) on LDL Oxidation in Vitro. Acta Med. Iran. 2013, 51, 12–18. [Google Scholar]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an Antioxidant and Disulphide Breaking Agent: The Reasons Why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeppilli, D.; Ribaudo, G.; Pompermaier, N.; Madabeni, A.; Bortoli, M.; Orian, L. Radical Scavenging Potential of Ginkgolides and Bilobalide: Insight from Molecular Modeling. Antioxidants 2023, 12, 525. [Google Scholar] [CrossRef] [PubMed]
- Njus, D.; Kelley, P.M.; Tu, Y.-J.; Schlegel, H.B. Ascorbic Acid: The Chemistry Underlying Its Antioxidant Properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, Y.; Zhan, L.; Rayat, G.R.; Xiao, J.; Jiang, H.; Li, X.; Chen, K. Anti-Diabetic Effects of Natural Antioxidants from Fruits. Trends Food Sci. Technol. 2021, 117, 3–14. [Google Scholar] [CrossRef]
- Zhao, C.-N.; Meng, X.; Li, Y.; Li, S.; Liu, Q.; Tang, G.-Y.; Li, H.-B. Fruits for Prevention and Treatment of Cardiovascular Diseases. Nutrients 2017, 9, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, A. Brain-Derived Neurotropic Factor/TrkB Signaling in the Pathogenesis and Novel Pharmacotherapy of Schizophrenia. Neurosignals 2008, 16, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.; Spencer, J.P.E.; Rice-Evans, C. Flavonoids: Antioxidants or Signalling Molecules? Free Radic. Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef]
- Ahmed, S.; Sulaiman, S.A.; Baig, A.A.; Ibrahim, M.; Liaqat, S.; Fatima, S.; Jabeen, S.; Shamim, N.; Othman, N.H. Honey as a Potential Natural Antioxidant Medicine: An Insight into Its Molecular Mechanisms of Action. Oxid. Med. Cell. Longev. 2018, 2018, 8367846. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.-H.; Chang, C.-S.; Ho, W.-C.; Liao, S.-Y.; Wu, C.-H.; Wang, C.-J. Anti-Metastasis Effects of Gallic Acid on Gastric Cancer Cells Involves Inhibition of NF-ΚB Activity and Downregulation of PI3K/AKT/Small GTPase Signals. Food Chem. Toxicol. 2010, 48, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Meshkibaf, M.H.; Maleknia, M.; Noroozi, S. Effect of Curcumin on Gene Expression and Protein Level of Methionine Sulfoxide Reductase A (MSRA), SOD, CAT and GPx in Freund’s Adjuvant Inflammation-Induced Male Rats. J. Inflamm. Res. 2019, 12, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Cornelius, C.; Trovato-Salinaro, A.; Cambria, M.; Locascio, M.; Rienzo, L.; Condorelli, D.; Mancuso, C.; De Lorenzo, A.; Calabrese, E. The Hormetic Role of Dietary Antioxidants in Free Radical-Related Diseases. Curr. Pharm. Des. 2010, 16, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Hrelia, S.; Angeloni, C. New Mechanisms of Action of Natural Antioxidants in Health and Disease. Antioxidants 2020, 9, 344. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Ikram, M.; Park, J.S.; Park, T.J.; Kim, M.O. Gut Microbiota, Its Role in Induction of Alzheimer’s Disease Pathology, and Possible Therapeutic Interventions: Special Focus on Anthocyanins. Cells 2020, 9, 853. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, G.; Madeddu, C.; Macciò, A.; Gramignano, G.; Lusso, M.R.; Massa, E.; Astara, G.; Serpe, R. Cancer-Related Anorexia/Cachexia Syndrome and Oxidative Stress: An Innovative Approach beyond Current Treatment. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1651–1659. [Google Scholar] [CrossRef]
- Shinn, L.J.; Lagalwar, S. Treating Neurodegenerative Disease with Antioxidants: Efficacy of the Bioactive Phenol Resveratrol and Mitochondrial-Targeted MitoQ and SkQ. Antioxidants 2021, 10, 573. [Google Scholar] [CrossRef]
- Speer, H.; D’Cunha, N.M.; Alexopoulos, N.I.; McKune, A.J.; Naumovski, N. Anthocyanins and Human Health—A Focus on Oxidative Stress, Inflammation and Disease. Antioxidants 2020, 9, 366. [Google Scholar] [CrossRef]
- Kabuto, H.; Amakawa, M.; Mankura, M.; Yamanushi, T.T.; Mori, A. Docosahexaenoic Acid Ethyl Ester Enhances 6-Hydroxydopamine-Induced Neuronal Damage by Induction of Lipid Peroxidation in Mouse Striatum. Neurochem. Res. 2009, 34, 1299–1303. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Klabnik, J.; O’ Donnell, J. Novel Therapeutic Targets in Depression and Anxiety: Antioxidants as a Candidate Treatment. Curr. Neuropharmacol. 2014, 12, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Ribaudo, G.; Bortoli, M.; Pavan, C.; Zagotto, G.; Orian, L. Antioxidant Potential of Psychotropic Drugs: From Clinical Evidence to In Vitro and In Vivo Assessment and toward a New Challenge for in Silico Molecular Design. Antioxidants 2020, 9, 714. [Google Scholar] [CrossRef]
- Ribaudo, G.; Bortoli, M.; Ongaro, A.; Oselladore, E.; Gianoncelli, A.; Zagotto, G.; Orian, L. Fluoxetine Scaffold to Design Tandem Molecular Antioxidants and Green Catalysts. RSC Adv. 2020, 10, 18583–18593. [Google Scholar] [CrossRef] [PubMed]
- Ribaudo, G.; Bortoli, M.; Witt, C.E.; Parke, B.; Mena, S.; Oselladore, E.; Zagotto, G.; Hashemi, P.; Orian, L. ROS-Scavenging Selenofluoxetine Derivatives Inhibit In Vivo Serotonin Reuptake. ACS Omega 2022, 7, 8314–8322. [Google Scholar] [CrossRef] [PubMed]
- Muraro, C.; Dalla Tiezza, M.; Pavan, C.; Ribaudo, G.; Zagotto, G.; Orian, L. Major Depressive Disorder and Oxidative Stress: In Silico Investigation of Fluoxetine Activity against ROS. Appl. Sci. 2019, 9, 3631. [Google Scholar] [CrossRef] [Green Version]
- Bortoli, M.; Dalla Tiezza, M.; Muraro, C.; Pavan, C.; Ribaudo, G.; Rodighiero, A.; Tubaro, C.; Zagotto, G.; Orian, L. Psychiatric Disorders and Oxidative Injury: Antioxidant Effects of Zolpidem Therapy Disclosed In Silico. Comput. Struct. Biotechnol. J. 2019, 17, 311–318. [Google Scholar] [CrossRef]
- sadat Yousefsani, B.; Akbarizadeh, N.; Pourahmad, J. The Antioxidant and Neuroprotective Effects of Zolpidem on Acrylamide-Induced Neurotoxicity Using Wistar Rat Primary Neuronal Cortical Culture. Toxicol. Rep. 2020, 7, 233–240. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current Trends in Computer Aided Drug Design and a Highlight of Drugs Discovered via Computational Techniques: A Review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Nimse, S.B.; Pal, D. Free Radicals, Natural Antioxidants, and Their Reaction Mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Orian, L.; Toppo, S. Organochalcogen Peroxidase Mimetics as Potential Drugs: A Long Story of a Promise Still Unfulfilled. Free Radic. Biol. Med. 2014, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- P Wolters, L.; Orian, L. Peroxidase Activity of Organic Selenides: Mechanistic Insights from Quantum Chemistry. Curr. Org. Chem. 2016, 20, 189–197. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Ribaudo, G.; Orian, L. Organodiselenides: Organic Catalysis and Drug Design Learning from Glutathione Peroxidase. Curr. Org. Chem. 2019, 23, 1381–1402. [Google Scholar] [CrossRef] [Green Version]
- Muraro, C.; Polato, M.; Bortoli, M.; Aiolli, F.; Orian, L. Radical Scavenging Activity of Natural Antioxidants and Drugs: Development of a Combined Machine Learning and Quantum Chemistry Protocol. J. Chem. Phys. 2020, 153, 114117. [Google Scholar] [CrossRef] [PubMed]
- Hernández-López, H.; Tejada-Rodríguez, C.J.; Leyva-Ramos, S. A Panoramic Review of Benzimidazole Derivatives and Their Potential Biological Activity. Mini Rev. Med. Chem. 2022, 22, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Anastassova, N.; Aluani, D.; Kostadinov, A.; Rangelov, M.; Todorova, N.; Hristova-Avakumova, N.; Argirova, M.; Lumov, N.; Kondeva-Burdina, M.; Tzankova, V.; et al. Evaluation of the Combined Activity of Benzimidazole Arylhydrazones as New Anti-Parkinsonian Agents: Monoamine Oxidase-B Inhibition, Neuroprotection and Oxidative Stress Modulation. Neural Regen. Res. 2021, 16, 2299–2309. [Google Scholar] [CrossRef]
- Imran, M.; Shah, F.A.; Nadeem, H.; Zeb, A.; Faheem, M.; Naz, S.; Bukhari, A.; Ali, T.; Li, S. Synthesis and Biological Evaluation of Benzimidazole Derivatives as Potential Neuroprotective Agents in an Ethanol-Induced Rodent Model. ACS Chem. Neurosci. 2021, 12, 489–505. [Google Scholar] [CrossRef]
- Ullah, A.; Al Kury, L.T.; Althobaiti, Y.S.; Ali, T.; Shah, F.A. Benzimidazole Derivatives as New Potential NLRP3 Inflammasome Inhibitors That Provide Neuroprotection in a Rodent Model of Neurodegeneration and Memory Impairment. J. Inflamm. Res. 2022, 15, 3873–3890. [Google Scholar] [CrossRef]
- Aleyasin, H.; Karuppagounder, S.S.; Kumar, A.; Sleiman, S.; Basso, M.; Ma, T.; Siddiq, A.; Chinta, S.J.; Brochier, C.; Langley, B.; et al. Antihelminthic Benzimidazoles Are Novel HIF Activators That Prevent Oxidative Neuronal Death via Binding to Tubulin. Antioxid. Redox Signal. 2015, 22, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Yadav, G.; Ganguly, S. Structure Activity Relationship (SAR) Study of Benzimidazole Scaffold for Different Biological Activities: A Mini-Review. Eur. J. Med. Chem. 2015, 97, 419–443. [Google Scholar] [CrossRef]
- Galano, A.; Reiter, R.J. Melatonin and Its Metabolites vs Oxidative Stress: From Individual Actions to Collective Protection. J. Pineal. Res. 2018, 65, e12514. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Raúl Alvarez-Idaboy, J. Computational Strategies for Predicting Free Radical Scavengers’ Protection against Oxidative Stress: Where Are We and What Might Follow? Int. J. Quantum Chem. 2019, 119, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Ribaudo, G.; Bellanda, M.; Menegazzo, I.; Wolters, L.P.; Bortoli, M.; Ferrer-Sueta, G.; Zagotto, G.; Orian, L. Mechanistic Insight into the Oxidation of Organic Phenylselenides by H2O2. Chem. Eur. J. 2017, 23, 2405–2422. [Google Scholar] [CrossRef]
- Ribaudo, G.; Bortoli, M.; Oselladore, E.; Ongaro, A.; Gianoncelli, A.; Zagotto, G.; Orian, L. Selenoxide Elimination Triggers Enamine Hydrolysis to Primary and Secondary Amines: A Combined Experimental and Theoretical Investigation. Molecules 2021, 26, 2770. [Google Scholar] [CrossRef]
- Teixeira Rocha, J.B.; Nogara, P.A.A.; Pereira, M.E.; Sirlene de Oliveira, C.; Orian, L. Organic Selenocompounds: Are They the Panacea to Human Illnesses? New J. Chem. 2023. [Google Scholar] [CrossRef]
- Ott, I. Metal N-Heterocyclic Carbene Complexes in Medicinal Chemistry. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2020; Volume 75, pp. 121–148. [Google Scholar]
- Tialiou, A.; Chin, J.; Keppler, B.K.; Reithofer, M.R. Current Developments of N-Heterocyclic Carbene Au(I)/Au(III) Complexes toward Cancer Treatment. Biomedicines 2022, 10, 1417. [Google Scholar] [CrossRef] [PubMed]
- Porchia, M.; Pellei, M.; Marinelli, M.; Tisato, F.; Del Bello, F.; Santini, C. New Insights in Au-NHCs Complexes as Anticancer Agents. Eur. J. Med. Chem. 2018, 146, 709–746. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Gimeno, M.C.; Visbal, R. Recent Advances in Gold–NHC Complexes with Biological Properties. Chem. Soc. Rev. 2019, 48, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Alberto, E.E.; Rossato, L.L.; Alves, S.H.; Alves, D.; Braga, A.L. Imidazolium Ionic Liquids Containing Selenium: Synthesis and Antimicrobial Activity. Org. Biomol. Chem. 2011, 9, 1001–1003. [Google Scholar] [CrossRef]
- Klauke, K.; Gruber, I.; Knedel, T.-O.; Schmolke, L.; Barthel, J.; Breitzke, H.; Buntkowsky, G.; Janiak, C. Silver, Gold, Palladium, and Platinum N-Heterocyclic Carbene Complexes Containing a Selenoether-Functionalized Imidazol-2-Ylidene Moiety. Organometallics 2018, 37, 298–308. [Google Scholar] [CrossRef]
- Sharma, K.N.; Joshi, H.; Sharma, A.K.; Prakash, O.; Singh, A.K. Selenium-Containing N-Heterocyclic Carbenes and Their First Palladium(II) Complexes: Synthesis, Structure, and Pendent Alkyl Chain Length Dependent Catalytic Activity for Suzuki–Miyaura Coupling. Organometallics 2013, 32, 2443–2451. [Google Scholar] [CrossRef]
- Bhaskar, R.; Sharma, A.K.; Singh, A.K. Palladium(II) Complexes of N-Heterocyclic Carbene Amidates Derived from Chalcogenated Acetamide-Functionalized 1 H-Benzimidazolium Salts: Recyclable Catalyst for Regioselective Arylation of Imidazoles under Aerobic Conditions. Organometallics 2018, 37, 2669–2681. [Google Scholar] [CrossRef]
- Klauke, K.; Zaitsau, D.H.; Bülow, M.; He, L.; Klopotowski, M.; Knedel, T.-O.; Barthel, J.; Held, C.; Verevkin, S.P.; Janiak, C. Thermodynamic Properties of Selenoether-Functionalized Ionic Liquids and Their Use for the Synthesis of Zinc Selenide Nanoparticles. Dalton Trans. 2018, 47, 5083–5097. [Google Scholar] [CrossRef]
- Dubey, P.; Singh, A.K. Sonogashira Coupling (Cu/Amine-Free) of ArBr/Cl in Aerobic Condition and N-Benzylation of Aniline with Benzyl Alcohol Catalyzed by Complexes of Pd(II) with Sulfated/Selenated NHCs. ChemistrySelect 2020, 5, 2925–2934. [Google Scholar] [CrossRef]
- Dubey, P.; Gupta, S.; Singh, A.K. Trinuclear Complexes of Palladium(II) with Chalcogenated N-Heterocyclic Carbenes: Catalysis of Selective Nitrile–Primary Amide Interconversion and Sonogashira Coupling. Dalton Trans. 2017, 46, 13065–13076. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Sharma, C.; Satrawala, N.; Srivastava, A.K.; Sharma, K.N.; Joshi, R.K. Selenium-Directed Ortho C–H Activation of Benzyl Selenide by a Selenated NHC–Half-Pincer Ruthenium(II) Complex. Organometallics 2022, 41, 1403–1411. [Google Scholar] [CrossRef]
- Khandaka, H.; Kumar Joshi, R. Fe3O4@SiO2Supported Pd (II)-Selenoether N-Heterocyclic Carbene: A Highly Active and Reusable Heterogeneous Catalyst for C O Cross-Coupling of Alcohols and Chloroarenes. Tetrahedron Lett. 2022, 111, 154163. [Google Scholar] [CrossRef]
- Tishchenko, O.; Truhlar, D.G.; Ceulemans, A.; Nguyen, M.T. A Unified Perspective on the Hydrogen Atom Transfer and Proton-Coupled Electron Transfer Mechanisms in Terms of Topographic Features of the Ground and Excited Potential Energy Surfaces As Exemplified by the Reaction between Phenol and Radicals. J. Am. Chem. Soc. 2008, 130, 7000–7010. [Google Scholar] [CrossRef] [PubMed]
- Draganić, I.G.; Draganićs, Z.D. The Radiation Chemistry of Water; Academic Press: New York, NY, USA, 1971; ISBN 9780323158787. [Google Scholar]
- Haber, F.; Weiss, J.; Seph, J.O.; Eiss, W. The Catalytic Decomposition of Hydrogen Peroxide by Iron Salts. Proc. Math. Phys. Sci. 1934, 147, 332–351. [Google Scholar] [CrossRef]
- Halliwell, B. Superoxide-Dependent Formation of Hydroxyl Radicals in the Presence of Iron Chelates. FEBS Lett. 1978, 92, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, E.; Gilles, M.K.; Ravishankara, A.R. Kinetics of the Reactions of the Hydroxyl Radical with CH3OH and C2H5OH between 235 and 360 K. J. Photochem. Photobiol. A Chem. 2003, 157, 237–245. [Google Scholar] [CrossRef]
- Pryor, W.A. Oxy-Radicals and Related Species: Their Formation, Lifetimes, and Reactions. Annu. Rev. Physiol. 1986, 48, 657–667. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Swart, M.; Bickelhaupt, F.M. Nucleophilic Substitution (SN2): Dependence on Nucleophile, Leaving Group, Central Atom, Substituents, and Solvent. ChemPhysChem 2018, 19, 1315–1330. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- ADF2016 SCM. Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2016; Available online: https://www.scm.com/ (accessed on 26 June 2023).
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A Density-Functional Model of the Dispersion Interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-Consistent Pseudopotentials for Group 11 and 12 Atoms: Adjustment to Multi-Configuration Dirac–Hartree–Fock Data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔGHAT, gas phase (kcal mol−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| C10.α | C10.β | C11.α | C11.β | C18.α | C18.β | C19.α | C19.β | C19.γ | |
| R | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| TS | 2.33 | 4.07 | −0.88 * | 5.43 | 3.55 | 1.40 | 3.48 | 5.71 | 0.83 |
| P | −20.98 | −20.97 | −19.57 | −19.57 | −21.59 | −24.3 | −16.4 | −16.55 | −16.6 |
| ΔGHAT, water (kcal mol−1) | |||||||||
| R | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| TS | 2.38 | 5.32 | 1.39 | 3.89 | 2.97 | 3.35 | 2.61 | 2.93 | 2.49 |
| P | −22.74 | −22.73 | −20.95 | −20.95 | −24.29 | −27.1 | −19.2 | −18.94 | −18.9 |
| ΔGHAT, benzene (kcal mol−1) | |||||||||
| R | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| TS | 2.32 | 4.69 | 0.16 | 4.59 | 3.53 | 2.34 | 3.31 | 4.40 | 1.93 |
| P | −21.39 | −21.38 | −19.32 | −19.32 | −22.06 | −24.62 | −16.64 | −16.39 | −16.38 |
| ΔGRAF (kcal mol−1) | |||
|---|---|---|---|
| Gas Phase | Water | Benzene | |
| R | 0.00 | 0.00 | 0.00 |
| TS | 1.17 | 1.63 | 2.21 |
| P | −23.17 | −19.13 | −19.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeppilli, D.; Aldinio-Colbachini, A.; Ribaudo, G.; Tubaro, C.; Dalla Tiezza, M.; Bortoli, M.; Zagotto, G.; Orian, L. Antioxidant Chimeric Molecules: Are Chemical Motifs Additive? The Case of a Selenium-Based Ligand. Int. J. Mol. Sci. 2023, 24, 11797. https://doi.org/10.3390/ijms241411797
Zeppilli D, Aldinio-Colbachini A, Ribaudo G, Tubaro C, Dalla Tiezza M, Bortoli M, Zagotto G, Orian L. Antioxidant Chimeric Molecules: Are Chemical Motifs Additive? The Case of a Selenium-Based Ligand. International Journal of Molecular Sciences. 2023; 24(14):11797. https://doi.org/10.3390/ijms241411797
Chicago/Turabian StyleZeppilli, Davide, Anna Aldinio-Colbachini, Giovanni Ribaudo, Cristina Tubaro, Marco Dalla Tiezza, Marco Bortoli, Giuseppe Zagotto, and Laura Orian. 2023. "Antioxidant Chimeric Molecules: Are Chemical Motifs Additive? The Case of a Selenium-Based Ligand" International Journal of Molecular Sciences 24, no. 14: 11797. https://doi.org/10.3390/ijms241411797
APA StyleZeppilli, D., Aldinio-Colbachini, A., Ribaudo, G., Tubaro, C., Dalla Tiezza, M., Bortoli, M., Zagotto, G., & Orian, L. (2023). Antioxidant Chimeric Molecules: Are Chemical Motifs Additive? The Case of a Selenium-Based Ligand. International Journal of Molecular Sciences, 24(14), 11797. https://doi.org/10.3390/ijms241411797