
Assessing the Link between Diabetic Metabolic Dysregulation and Breast Cancer Progression

 , , , ,
, , , , 
Abstract
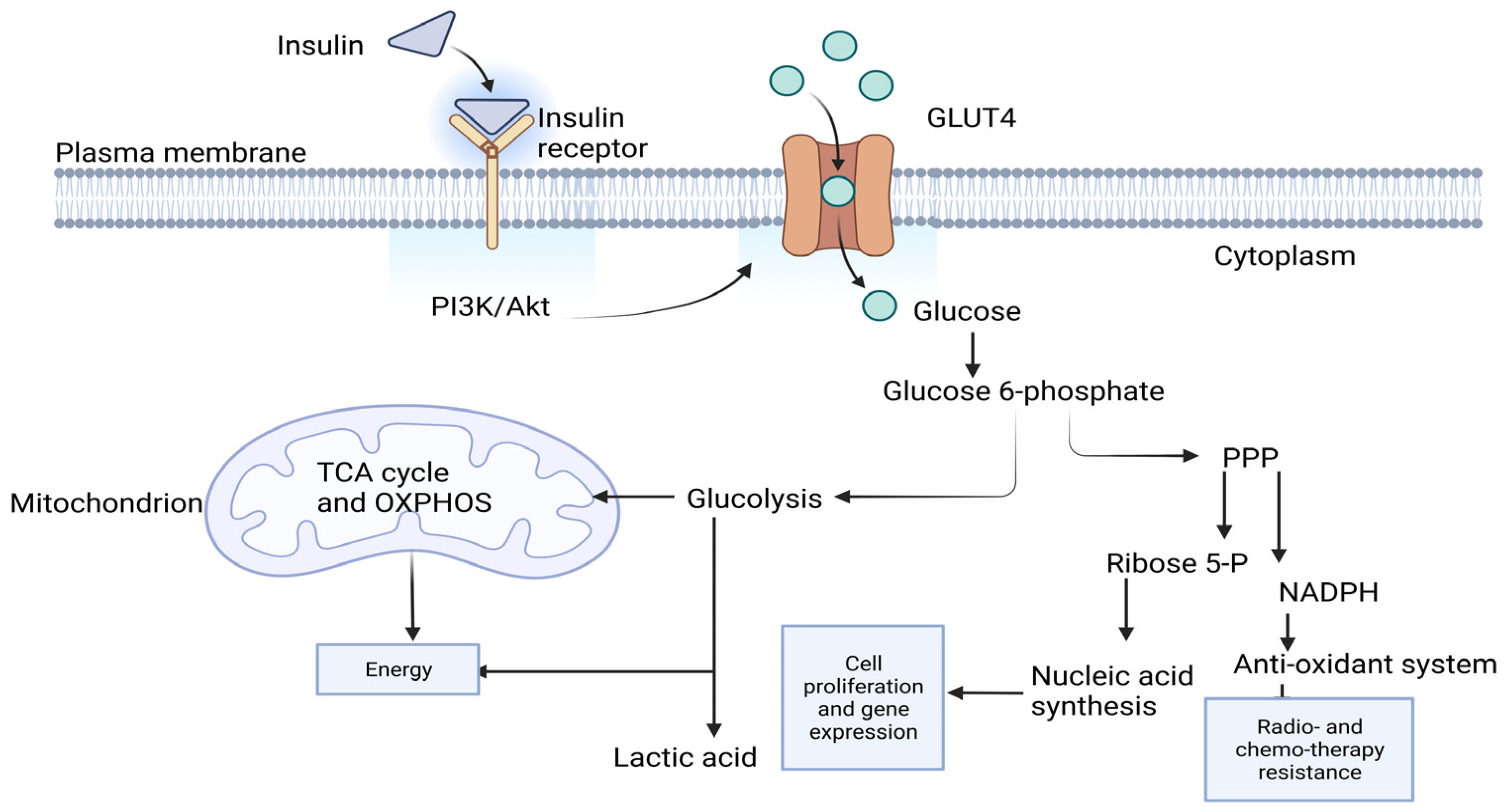
:1. Introduction
2. Results
2.1. Differentially Expressed Genes in MCF7 with Different Treatment
2.2. The Overlapping Differentially Expressed Genes from In Vitro Experiments and In Silico Data
3. Discussion
4. Materials and Methods
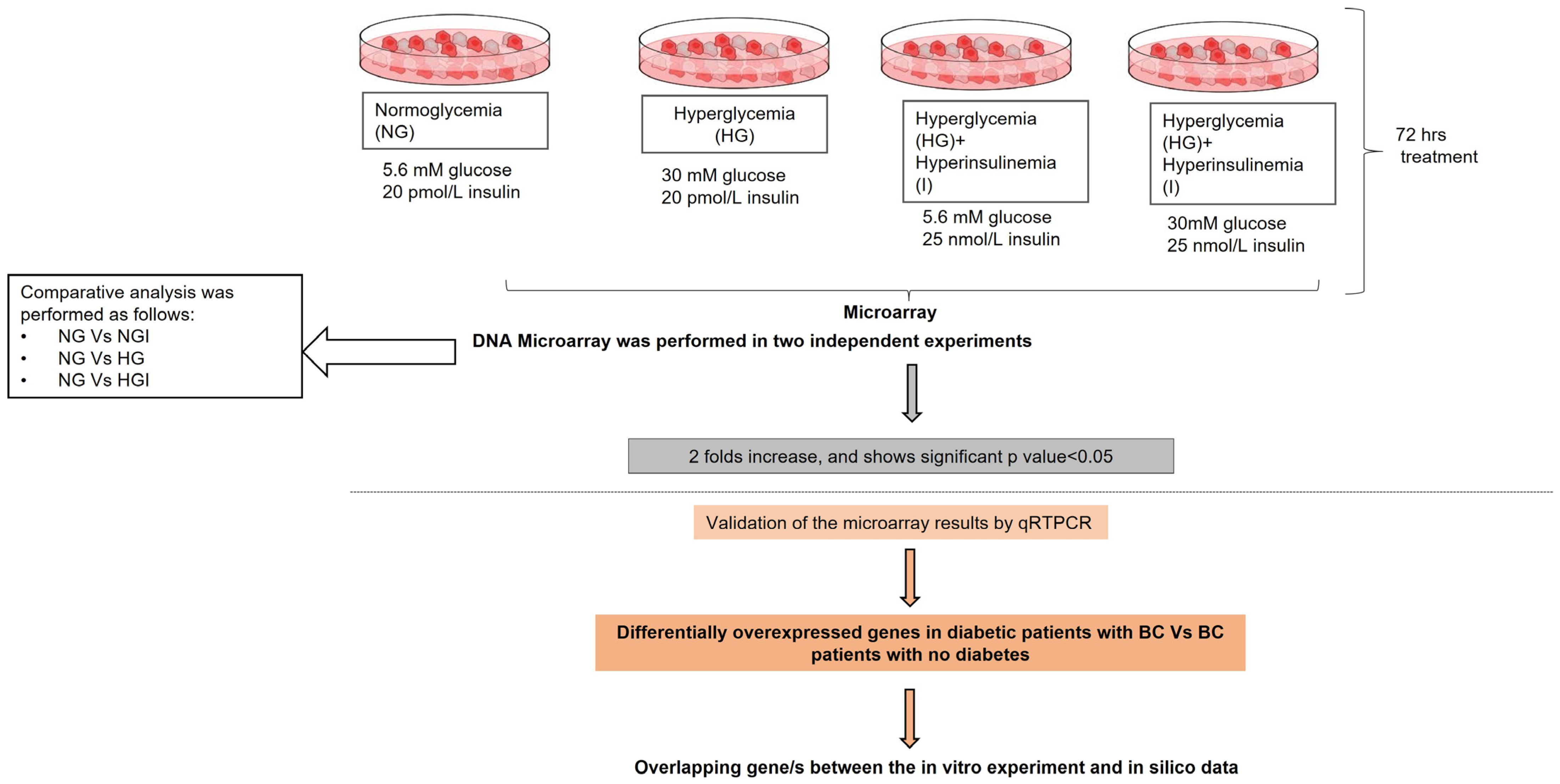
4.1. Study Design
4.2. Deciding Glucose and Insulin Levels
4.3. Cell Culture
4.4. RNA Extraction
4.5. RNA Quality Control
4.6. Labelling and Microarray Hybridization
4.7. Microarray Data Analysis
4.8. In Silico Analysis
4.9. RT-PCR
4.10. Statistical Analysis
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Q.-L.; Xu, W.-H.; Tao, M.-H. Biomarkers of the Metabolic Syndrome and Breast Cancer Prognosis. Cancers 2010, 2, 721–739. [Google Scholar] [CrossRef]
- Lega, I.C.; Austin, P.C.; Fischer, H.D.; Fung, K.; Krzyzanowska, M.K.; Amir, E.; Lipscombe, L.L. The Impact of Diabetes on Breast Cancer Treatments and Outcomes: A Population-Based Study. Diabetes Care 2018, 41, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.B.; Ren, G.S. Diabetes mellitus and prognosis in women with breast cancer: A systematic review and meta-analysis. Medicine 2016, 95, e5602. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.J.; Kendall, C.W.; Augustin, L.S.; Franceschi, S.; Hamidi, M.; Marchie, A.; Jenkins, A.L.; Axelsen, M. Glycemic index: Overview of implications in health and disease. Am. J. Clin. Nutr. 2002, 76, 266S–273S. [Google Scholar] [CrossRef]
- Belle, F.N.; Kampman, E.; McTiernan, A.; Bernstein, L.; Baumgartner, K.; Baumgartner, R.; Ambs, A.; Ballard-Barbash, R.; Neuhouser, M.L. Dietary Fiber, Carbohydrates, Glycemic Index, and Glycemic Load in Relation to Breast Cancer Prognosis in the HEAL Cohort. Cancer Epidemiol. Biomark. Prev. 2011, 20, 890–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farvid, M.S.; Tamimi, R.M.; Poole, E.M.; Chen, W.Y.; Rosner, B.A.; Willett, W.C.; Holmes, M.D.; Eliassen, A.H. Postdiagnostic Dietary Glycemic Index, Glycemic Load, Dietary Insulin Index, and Insulin Load and Breast Cancer Survival. Cancer Epidemiol. Biomark. Prev. 2021, 30, 335–343. [Google Scholar] [CrossRef]
- Garrido, P.; Osorio, F.G.; Morán, J.; Cabello, E.; Alonso, A.; Freije, J.M.; González, C. Loss of GLUT4 Induces Metabolic Reprogramming and Impairs Viability of Breast Cancer Cells. J. Cell. Physiol. 2015, 230, 191–198. [Google Scholar] [CrossRef]
- Chang, L.; Chiang, S.-H.; Saltiel, A.R. Insulin Signaling and the Regulation of Glucose Transport. Mol. Med. 2004, 10, 65–71. [Google Scholar] [CrossRef]
- Patel, J.H.; Ong, D.J.; Williams, C.R.; Callies, L.K.; Wills, A.E. Elevated pentose phosphate pathway flux supports appendage regeneration. Cell Rep. 2022, 41, 111552. [Google Scholar] [CrossRef]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xia, L.; Liang, J.; Han, Y.; Wang, H.; Oyang, L.; Tan, S.; Tian, Y.; Rao, S.; Chen, X.; et al. The roles of glucose metabolic reprogramming in chemo- and radio-resistance. J. Exp. Clin. Cancer Res. 2019, 38, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denga, H.; Chena, Y.; Lia, P.; Hanga, Q.; Zhanga, P.; Jina, Y.; Chen, M. PI3K/AKT/mTOR pathway, hypoxia, and glucose metabolism: Potential targets to overcome radioresistance in small cell lung cancer. Cancer Pathog. Ther. 2023, 1, 56–66. [Google Scholar] [CrossRef]
- Hou, Y.; Zhou, M.; Xie, J.; Chao, P.; Feng, Q.; Wu, J. High glucose levels promote the proliferation of breast cancer cells through GTPases. Breast Cancer Targets Ther. 2017, 9, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Ma, Q.; Li, J.; Liu, H.; Li, W.; Ma, G.; Xu, Q.; Zhou, S.; Wu, E. High Glucose Promotes Pancreatic Cancer Cell Proliferation via the Induction of EGF Expression and Transactivation of EGFR. PLoS ONE 2011, 6, e27074. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Arumugam, A.; Joseph, R.; Monga, K.; Boopalan, T.; Agullo, P.; Gutierrez, C.; Nandy, S.; Subramani, R.; de la Rosa, J.M.; et al. Hyperglycemia Enhances the Proliferation of Non-Tumorigenic and Malignant Mammary Epithelial Cells through Increased leptin/IGF1R Signaling and Activation of AKT/mTOR. PLoS ONE 2013, 8, e79708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-López, L.A.; Martínez-Hernández, M.G.; Viedma-Rodríguez, R.; Díaz-Flores, M.; Baiza-Gutman, L.A. High glucose and insulin enhance uPA expression, ROS formation and invasiveness in breast cancer-derived cells. Cell. Oncol. 2016, 39, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Fainsod-Levi, T.; Gershkovitz, M.; Völs, S.; Kumar, S.; Khawaled, S.; Sagiv, J.Y.; Sionov, R.V.; Grunewald, M.; Keshet, E.; Granot, Z. Hyperglycemia Impairs Neutrophil Mobilization Leading to Enhanced Metastatic Seeding. Cell Rep. 2017, 21, 2384–2392. [Google Scholar] [CrossRef] [Green Version]
- Tomaskovic-Crook, E.; Thompson, E.W.; Thiery, J.P. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 2009, 11, 213. [Google Scholar] [CrossRef]
- Li, W.; Ma, Z.; Ma, J.; Li, X.; Xu, Q.; Duan, W.; Chen, X.; Lv, Y.; Zhou, S.; Wu, E.; et al. Hydrogen peroxide mediates hyperglycemia-induced invasive activity via ERK and p38 MAPK in human pancreatic cancer. Oncotarget 2015, 6, 31119–31133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Chen, R.; Zhao, M.; Li, L.; Fan, L.; Che, X.-M. High glucose promotes gastric cancer chemoresistance in vivo and in vitro. Mol. Med. Rep. 2015, 12, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- anssen, J.A.M.J.L. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef]
- Gupta, C.; Tikoo, K. High glucose and insulin differentially modulates proliferation in MCF-7 and MDA-MB-231 cells. J. Mol. Endocrinol. 2013, 51, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.-L.; Duan, P.; Wang, Z.-M.; Ding, M.; Tu, P. High glucose and high insulin conditions promote MCF-7 cell proliferation and invasion by upregulating IRS1 and activating the Ras/Raf/ERK pathway. Mol. Med. Rep. 2017, 16, 6690–6696. [Google Scholar] [CrossRef] [Green Version]
- WHO. Breast Cancer; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef] [Green Version]
- Kapur, A.; Seshiah, V. Women & diabetes: Our right to a healthy future. Indian J. Med. Res. 2017, 146, 553–556. [Google Scholar] [CrossRef]
- Calculate and Draw Custom Venn Diagrams Bioinformatics & Evolutionary Genomics: Van de Peer Lab. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 4 December 2022).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, K.; Stuckey, A. Breast Cancer Epidemiology and Risk Factors. Clin. Obstet. Gynecol. 2016, 59, 651–672. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Narayan, S.; Sharma, N.; Sharma, R.; Kapoor, A.; Kumar, N.; Nirban, R. Association of comorbidities with breast cancer: An observational study. Trop. J. Med. Res. 2015, 19, 168. [Google Scholar] [CrossRef] [Green Version]
- Ewertz, M.; Land, L.H.; Dalton, S.O.; Cronin-Fenton, D.; Jensen, M.-B. Influence of specific comorbidities on survival after early-stage breast cancer. Acta Oncol. 2018, 57, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgel, E.; Mittelman, S.D. The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altundag, K. Breast Cancer Molecular Subtypes and Chemotherapy Schedules Used in Neoadjuvant or Adjuvant Setting May Show Different Effects in Nipple-Sparing Mastectomy. Plast. Reconstr. Surg. 2017, 140, 495e. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, W.; Huh, S.J.; Choi, D.H.; Noh, J.M.; Im, Y.-H.; Ahn, J.S.; Park, Y.H.; Nam, S.J.; Kim, S.W.; et al. Clinical outcomes according to molecular subtypes in stage II-III breast cancer patients treated with neoadjuvant chemotherapy followed by surgery and radiotherapy. Asia-Pacific J. Clin. Oncol. 2017, 13, 329–336. [Google Scholar] [CrossRef]
- Subbiah, S.; Gopu, G.; Senthilkumar, P.; Muniasamy, P. Molecular subtypes as a predictor of response to neoadjuvant chemotherapy in breast cancer patients. Indian J. Cancer 2017, 54, 652–657. [Google Scholar] [CrossRef]
- Nilsson, P.M.; Tuomilehto, J.; Ryden, L. The metabolic syndrome—What is it and how should it be managed? Eur. J. Prev. Cardiol. 2019, 26, 33–46. [Google Scholar] [CrossRef]
- Martyn, J.A.; Kaneki, M.; Yasuhara, S. Obesity-induced insulin resistance and hyperglycemia: Etiologic factors and molecular mechanisms. Anesthesiology 2008, 109, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Barbot, M.; Ceccato, F.; Scaroni, C. Diabetes Mellitus Secondary to Cushing’s Disease. Front. Endocrinol. 2018, 9, 284. [Google Scholar] [CrossRef]
- Wintergerst, K.A.; Rogers, E.S.; Foster, M.B. Hyperthyroidism presenting with hyperglycemia in an adolescent female. J. Pediatr. Endocrinol. Metab. 2011, 24, 385–387. [Google Scholar] [CrossRef]
- Hu, Y.; Gao, G.; Yan, R.-N.; Li, F.-F.; Su, X.-F.; Ma, J.-H. Glucose metabolism before and after radioiodine therapy of a patient with Graves’ disease: Assessment by continuous glucose monitoring. Biomed. Rep. 2017, 7, 183–187. [Google Scholar] [CrossRef] [Green Version]
- McCowen, K.C.; Malhotra, A.; Bistrian, B.R. Stress-induced hyperglycemia. Crit. Care Clin. 2001, 17, 107–124. [Google Scholar] [CrossRef]
- Gregoriou, K.; Craigie, I.; Gibson, B.; Mason, A.; Shaikh, M.G. Risk factors and management of corticosteroid-induced hyperglycaemia in paediatric acute lymphoblastic leukaemia. Pediatr. Blood Cancer 2020, 67, e28085. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-P.; Teng, Y.-C.; Zhou, J.; Lu, W.; Tao, M.-F.; Jia, W.-P. Increased mean glucose levels in patients with polycystic ovary syndrome and hyperandrogenemia as determined by continuous glucose monitoring. Acta Obstet. Gynecol. Scand. 2013, 92, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Dumais, V.; Lumingu, J.; Bedard, M.; Paquet, L.; Verma, S.; Fontaine-Bisson, B. Prevalence of Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes in Canadian Women at High Risk for Breast Cancer. Breast J. 2017, 23, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int. J. Cancer 2017, 141, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, B.; Aziz, S.A.; Ganaie, M.A.; Mir, M.H. Metabolic syndrome and breast cancer risk. Indian J. Med. Paediatr. Oncol. 2017, 38, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Porto, L.A.; Lora, K.J.B.; Soares, J.C.M.; Costa, L.O.B.F. Metabolic syndrome is an independent risk factor for breast cancer. Arch. Gynecol. Obstet. 2011, 284, 1271–1276. [Google Scholar] [CrossRef]
- Eketunde, A.O. Diabetes as a Risk Factor for Breast Cancer. Cureus 2020, 12, e8010. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Annibaldi, A.; Widmann, C. Glucose metabolism in cancer cells. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 466–470. [Google Scholar] [CrossRef]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-Induced Reactive Oxygen Species Increase Expression of the Receptor for Advanced Glycation End Products (RAGE) and RAGE Ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novosyadlyy, R.; Lann, D.E.; Vijayakumar, A.; Rowzee, A.; Lazzarino, D.A.; Fierz, Y.; Carboni, J.M.; Gottardis, M.M.; Pennisi, P.A.; Molinolo, A.A.; et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 2010, 70, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Heuson, J.C.; Legros, N.; Heimann, R. Influence of insulin administration on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in intact, oophorectomized, and hypophysectomized rats. Cancer Res. 1972, 32, 233–238. [Google Scholar] [PubMed]
- Corpet, D.E.; Jacquinet, C.; Peiffer, G.; Taché, S. Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutr. Cancer 1997, 27, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Bates, S.E.; Davidson, N.E.; Valverius, E.M.; Freter, C.E.; Dickson, R.B.; Tam, J.P.; Kudlow, J.E.; Lippman, M.E.; Salomon, D.S. Expression of Transforming Growth Factor α and its Messenger Ribonucleic Acid in Human Breast Cancer: Its Regulation by Estrogen and its Possible Functional Significance. Mol. Endocrinol. 1988, 2, 543–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Cui, H.; Gao, Y.; Pan, Y.; Zhou, K.; Huang, J.; Lan, J.; Wei, Q.; Liu, X.; Liu, L.; et al. TGF-α Overexpression in Breast Cancer Bone Metastasis and Primary Lesions and TGF-α Enhancement of Expression of Procancer Metastasis Cytokines in Bone Marrow Mesenchymal Stem Cells. BioMed Res. Int. 2018, 2018, 6565393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, D.A.; Paterson, A.J.; Roos, M.D.; Wei, X.; Kudlow, J.E. Glucose and glucosamine regulate growth factor gene expression in vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 1992, 89, 8150–8154. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Liu, Y.; Zhang, Y.; Fu, Q.; Liu, Z.; Wang, Z.; Fu, H.; Xu, J.; Liu, K. Increased expression of MUC3A is associated with poor prognosis in localized clear-cell renal cell carcinoma. Oncotarget 2016, 7, 50017–50026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, C.; Zha, H.; Long, H.; Wang, X.; Yang, F.; Gao, J.; Hu, C.; Zhou, L.; Guo, B.; Zhu, B. C3a-C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J. Exp. Clin. Cancer Res. 2020, 39, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xia, C.; Pu, M.; Dai, B.; Yang, X.; Shang, R.; Yang, Z.; Zhang, R.; Tao, K.; Dou, K. Silencing of CDCA5 inhibits cancer progression and serves as a prognostic biomarker for hepatocellular carcinoma. Oncol. Rep. 2018, 40, 1875–1884. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Koinuma, J.; Ueda, K.; Ito, T.; Tsuchiya, E.; Nakamura, Y.; Daigo, Y. Phosphorylation and activation of cell division cycle associated 5 by mitogen-activated protein kinase play a crucial role in human lung carcinogenesis. Cancer Res. 2010, 70, 5337–5347. [Google Scholar] [CrossRef] [Green Version]
- Phan, N.N.; Wang, C.-Y.; Li, K.-L.; Chen, C.-F.; Chiao, C.-C.; Yu, H.-G.; Huang, P.-L.; Lin, Y.-C. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget 2018, 9, 6977–6992. [Google Scholar] [CrossRef]
- C3AR1. Available online: https://www.proteinatlas.org/ENSG00000171860-C3AR1/pathology/renal+cancer (accessed on 3 June 2021).
- Ruokun, C.; Yake, X.; Fengdong, Y.; Xinting, W.; Laijun, S.; Xianzhi, L. Lentivirus-mediated silencing of HSDL2 suppresses cell proliferation in human gliomas. Tumor Biol. 2016, 37, 15065–15077. [Google Scholar] [CrossRef]
- Weber, H.; Garabedian, M.J. The mediator complex in genomic and non-genomic signaling in cancer. Steroids 2018, 133, 8–14. [Google Scholar] [CrossRef]
- Leonard, M.; Zhang, X. Estrogen receptor coactivator Mediator Subunit 1 (MED1) as a tissue-specific therapeutic target in breast cancer. J. Zhejiang Univ. B 2019, 20, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Irshad, S.; Yu, W.; Belakavadi, M.; Chekmareva, M.; Ittmann, M.M.; Abate-Shen, C.; Fondell, J.D. ERK and AKT signaling drive MED1 overexpression in prostate cancer in association with elevated proliferation and tumorigenicity. Mol. Cancer Res. 2013, 11, 736–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Reymúndez, A.; Vázquez, A.I. Multi-omic signatures identify pan-cancer classes of tumors beyond tissue of origin. Sci. Rep. 2020, 10, 8341. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yin, B.; Wang, Q.; Ju, W.; Chen, Y.; Qiu, H.; Li, J.; Peng, X.; Lu, C. Cytoplasmic Poly(A) Binding Protein 4 Is Highly Expressed in Human Colorectal Cancer and Correlates with Better Prognosis. J. Genet. Genom. 2012, 39, 369–374. [Google Scholar] [CrossRef]
- PABPC4. Available online: https://www.proteinatlas.org/ENSG00000090621-PABPC4/pathology (accessed on 3 June 2021).
- Pequerul, R.; Vera, J.; Giménez-Dejoz, J.; Crespo, I.; Coines, J.; Porté, S.; Rovira, C.; Parés, X.; Farrés, J. Structural and kinetic features of aldehyde dehydrogenase 1A (ALDH1A) subfamily members, cancer stem cell markers active in retinoic acid biosynthesis. Arch. Biochem. Biophys. 2020, 681, 108256. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Liu, R.-Z.; Coyle, K.M.; Bydoun, M.; Wallace, M.; Clements, D.; Turner, C.; Mathenge, E.G.; Gujar, S.A.; et al. Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol. Oncol. 2015, 9, 17–31. [Google Scholar] [CrossRef]
- Yao, J.; Cui, Q.; Fan, W.; Ma, Y.; Chen, Y.; Liu, T.; Zhang, X.; Xi, Y.; Wang, C.; Peng, L.; et al. Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer. Nat. Commun. 2020, 11, 3715. [Google Scholar] [CrossRef]
- Nie, S.; Qian, X.; Shi, M.; Li, H.; Peng, C.; Ding, X.; Zhang, S.; Zhang, B.; Xu, G.; Lv, Y.; et al. ALDH1A3 Accelerates Pancreatic Cancer Metastasis by Promoting Glucose Metabolism. Front. Oncol. 2020, 10, 915. [Google Scholar] [CrossRef]
- Kawakami, R.; Mashima, T.; Kawata, N.; Kumagai, K.; Migita, T.; Sano, T.; Mizunuma, N.; Yamaguchi, K.; Seimiya, H. ALDH1A3-mTOR axis as a therapeutic target for anticancer drug-tolerant persister cells in gastric cancer. Cancer Sci. 2020, 111, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde Dehydrogenase Activity of Breast Cancer Stem Cells Is Primarily Due To Isoform ALDH1A3 and Its Expression Is Predictive of Metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef]
- Yamashita, D.; Minata, M.; Ibrahim, A.N.; Yamaguchi, S.; Coviello, V.; Bernstock, J.D.; Harada, S.; Cerione, R.A.; Tannous, B.A.; La Motta, C.; et al. Identification of ALDH1A3 as a Viable Therapeutic Target in Breast Cancer Metastasis-Initiating Cells. Mol. Cancer Ther. 2020, 19, 1134–1147. [Google Scholar] [CrossRef] [Green Version]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019, 26, 3061–3075.e6. [Google Scholar] [CrossRef] [Green Version]
- Gelardi, E.L.M.; Colombo, G.; Picarazzi, F.; Ferraris, D.M.; Mangione, A.; Petrarolo, G.; Aronica, E.; Rizzi, M.; Mori, M.; La Motta, C.; et al. A Selective Competitive Inhibitor of Aldehyde Dehydrogenase 1A3 Hinders Cancer Cell Growth, Invasiveness and Stemness In Vitro. Cancers 2021, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, N.; Kunzelmann, K.; Xu, J.; Barone, S.; Sirianant, L.; De Zeeuw, C.I.; Soleimani, M. Slc26a11 is prominently expressed in the brain and functions as a chloride channel: Expression in Purkinje cells and stimulation of V H+-ATPase. Pflug. Arch. 2013, 465, 1583–1597. [Google Scholar] [CrossRef]
- Vincourt, J.B.; Jullien, D.; Amalric, F.; Girard, J.P. Molecular and functional characterization of SLC26A11, a sodium-independent sulfate transporter from high endothelial venules. FASEB J. 2003, 17, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Barone, S.; Li, H.; Holiday, S.; Zahedi, K.; Soleimani, M. Slc26a11, a chloride transporter, localizes with the vacuolar H(+)-ATPase of A-intercalated cells of the kidney. Kidney Int. 2011, 80, 926–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhang, C.; Liu, H.; Xu, C.; Duan, H.; Tian, X.; Zhang, D. Heritability and genome-wide association analyses of fasting plasma glucose in Chinese adult twins. BMC Genom. 2020, 21, 491. [Google Scholar] [CrossRef]
- Zhu, J.; Mou, Y.; Ye, S.; Hu, H.; Wang, R.; Yang, Q.; Hu, Y. Identification of a Six-Gene SLC Family Signature With Prognostic Value in Patients With Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 803198. [Google Scholar] [CrossRef]
- SLC26A11. Available online: https://www.proteinatlas.org/ENSG00000181045-SLC26A11/pathology (accessed on 4 December 2022).
- Zhou, J.-B.; Zhang, T.; Wang, B.-F.; Gao, H.-Z.; Xu, X. Identification of a novel gene fusion RNF213-SLC26A11 in chronic myeloid leukemia by RNA-Seq. Mol. Med. Rep. 2013, 7, 591–597. [Google Scholar] [CrossRef] [Green Version]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Faculty Opinions recommendation of Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Senapati, P.; Kato, H.; Lee, M.; Leung, A.; Thai, C.; Sanchez, A.; Gallagher, E.J.; LeRoith, D.; Seewaldt, V.L.; Ann, D.K.; et al. Hyperinsulinemia promotes aberrant histone acetylation in triple-negative breast cancer. Epigenet. Chromatin 2019, 12, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nankali, M.; Karimi, J.; Goodarzi, M.T.; Saidijam, M.; Khodadadi, I.; Emami Razavi, A.N.; Rahimi, F. Increased Expression of the Receptor for Advanced Glycation End-Products (RAGE) Is Associated with Advanced Breast Cancer Stage. Oncol Res Treat 2016, 39, 622–628. [Google Scholar] [CrossRef]
- Fuentes, M.K.; Nigavekar, S.S.; Arumugam, T.; Logsdon, C.D.; Schmidt, A.M.; Park, J.C.; Huang, E.H. RAGE Activation by S100P in Colon Cancer Stimulates Growth, Migration, and Cell Signaling Pathways. Dis. Colon Rectum 2007, 50, 1230–1240. [Google Scholar] [CrossRef]
- Arumugam, T.; Simeone, D.M.; Schmidt, A.M.; Logsdon, C.D. S100P Stimulates Cell Proliferation and Survival via Receptor for Activated Glycation End Products (RAGE). J. Biol. Chem. 2004, 279, 5059–5065. [Google Scholar] [CrossRef] [Green Version]
- SPINK8. Available online: https://www.proteinatlas.org/ENSG00000229453-SPINK8/pathology (accessed on 3 June 2021).
- Esper, A.M.; Moss, M.; Martin, G.S. The effect of diabetes mellitus on organ dysfunction with sepsis: An epidemiological study. Crit. Care 2009, 13, R18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Comşa, Ş.; Cîmpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res. 2015, 35, 3147–3154. [Google Scholar]
- Sarvaiya, H.A.; Lazar, I.M. Insulin stimulated MCF7 breast cancer cells: Proteome dataset. Data Brief 2016, 9, 579–584. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Milazzo, G.; Giorgino, F.; Damante, G.; Sung, C.; Stampfer, M.R.; Vigneri, R.; Goldfine, I.D.; Belfiore, A. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res. 1992, 52, 3924–3930. [Google Scholar] [PubMed]
- Li, W.; Zhang, X.; Sang, H.; Zhou, Y.; Shang, C.; Wang, Y.; Zhu, H. Effects of hyperglycemia on the progression of tumor diseases. J. Exp. Clin. Cancer Res. 2019, 38, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.M.; Hussain, F. Higher Glucose Enhances Breast Cancer Cell Aggressiveness. Nutr. Cancer 2020, 72, 734–746. [Google Scholar] [CrossRef]
- World Health Organization & International Diabetes Federation. Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycaemia: Report of a WHO/IDF Consultation. World Health Organization. 2006. Available online: https://apps.who.int/iris/handle/10665/43588 (accessed on 19 July 2023).
- Insulin. 2019. Available online: https://www.medscape.com/answers/2089224-170937/what-are-the-reference-ranges-of-insulin-levels (accessed on 11 January 2022).
- Shlomo Melmed, K.S.P.; Reed Larsen, P.; Kronenberg, H.M. Williams Textbook of Endocrinology, 13th ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Wallach, J. Wallach’s Interpretation of Diagnostic Tests: Pathways to Arriving at a Clinical Diagnosis, 10th ed.; Williamson, M.A., Michael Synder, L., Eds.; Wolters Kluwer: Tokyo, Japan, 2015. [Google Scholar]
- Govindaraj, L.; Gupta, T.; Esvaran, V.G.; Awasthi, A.K.; Ponnuvel, K.M. Genome-wide identification, characterization of sugar transporter genes in the silkworm Bombyx mori and role in Bombyx mori nucleopolyhedrovirus (BmNPV) infection. Gene 2016, 579, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; He, Q. Next Generation Sequencing Identifies Differentially Expressed Genes between Breast Cancer with Diabetes and Breast Cancer without Diabetes [GEO]. NCBI. 2020. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150586 (accessed on 18 November 2020).
- Rachinger, N.; Fischer, S.; Böhme, I.; Linck-Paulus, L.; Kuphal, S.; Kappelmann-Fenzl, M.; Bosserhoff, A.K. Loss of Gene Information: Discrepancies between RNA Sequencing, cDNA Microarray, and qRT-PCR. Int. J. Mol. Sci. 2021, 22, 9349. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, P.; Yarani, R.; Valipour, E.; Kiani, S.; Hoseinkhani, Z.; Mansouri, K. Cell line-directed breast cancer research based on glucose metabolism status. Biomed. Pharmacother. 2022, 146, 112526. [Google Scholar] [CrossRef] [PubMed]
- Gardner, G.L.; Moradi, F.; Moffatt, C.; Cliche, M.; Garlisi, B.; Gratton, J.; Mehmood, F.; Stuart, J.A. Rapid nutrient depletion to below the physiological range by cancer cells cultured in Plasmax. Am. J. Physiol. Physiol. 2022, 323, C823–C834. [Google Scholar] [CrossRef]
- Al-Ani, A.; Toms, D.; Kondro, D.; Thundathil, J.; Yu, Y.; Ungrin, M. Oxygenation in cell culture: Critical parameters for reproducibility are routinely not reported. PLoS ONE 2018, 13, e0204269. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Zhang, W.; Yuan, Q.; Niu, M.; Zhou, F.; Qiu, Q.; Mao, G.; Wang, H.; Wen, L.; Sun, M.; et al. PAK1 Promotes the Proliferation and Inhibits Apoptosis of Human Spermatogonial Stem Cells via PDK1/KDR/ZNF367 and ERK1/2 and AKT Pathways. Mol. Ther. Nucleic Acids. 2018, 12, 769–786. [Google Scholar] [CrossRef]
- Sullivan, K.E.; Rojas, K.; Cerione, R.A.; Nakano, I.; Wilson, K.F. The stem cell/cancer stem cell marker ALDH1A3 regulates the expression of the survival factor tissue transglutaminase, in mesenchymal glioma stem cells. Oncotarget 2017, 8, 22325–22343. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, D.C.; L’Hôte, C.G.; Kennedy, W.; Pitt, E.; Knowles, M.A. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Res. 2005, 65, 10441–10449. [Google Scholar] [CrossRef] [Green Version]
- Tokuzen, N.; Nakashiro, K.; Tanaka, H.; Iwamoto, K.; Hamakawa, H. Therapeutic potential of targeting cell division cycle associated 5 for oral squamous cell carcinoma. Oncotarget 2016, 7, 2343–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, J.R.; Fuzii, H.T.; Kayser, C.; Alberto, F.L.; Soares, F.A.; Sato, E.I.; Andrade, L.E. IL-2, IL-5, TNF-α and IFN-γ mRNA expression in epidermal keratinocytes of systemic lupus erythematosus skin lesions. Clinics 2011, 66, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Average of Fold Change | p Value < 0.05 | Adj-p-Value | Description |
|---|---|---|---|---|
| SNRPC | 5.613 | <0.001 | 0.006 | Small nuclear ribonucleoprotein polypeptide C |
| FGFR3 | 3.071 | <0.001 | 0.006 | Fibroblast growth factor receptor 3 |
| FAM219A | 3.469 | <0.001 | 0.005 | Family with sequence similarity 219, member A |
| SLC26A11 | 4.311 | <0.001 | 0.005 | Solute carrier family 26 (anion exchanger), member 11 |
| ATMIN | 2.055 | <0.001 | 0.004 | ATM interactor |
| ARHGEF10 | 3.419 | <0.001 | 0.004 | Rho guanine nucleotide exchange factor (GEF) 10 |
| PAK1 | 2.131 | <0.001 | 0.004 | p21 protein (Cdc42/Rac)-activated kinase 1 |
| JPH3 | 5.196 | <0.001 | 0.005 | Junctophilin 3 |
| BRD7 | 4.668 | <0.001 | 0.005 | Bromodomain containing 7 |
| GNB5 | 2.382 | <0.001 | 0.006 | Guanine nucleotide binding protein (G protein), beta 5 |
| LEPR | 5.740 | <0.001 | 0.005 | Leptin receptor |
| TMEM180 | 3.148 | <0.001 | 0.005 | Transmembrane protein 180 |
| SCP2 | 3.759 | <0.001 | 0.005 | Sterol carrier protein 2 |
| CDCA5 | 4.805 | <0.001 | 0.005 | Cell division cycle associated 5 |
| DYNLT1 | 5.364 | 0.001 | 0.005 | Dynein, light chain, Tctex-type 1 |
| RBM14 | 2.333 | 0.001 | 0.005 | RNA binding motif protein 14 |
| GNAZ | 3.279 | 0.001 | 0.006 | Guanine nucleotide binding protein (G protein), alpha z polypeptide |
| STAC3 | 2.354 | 0.001 | 0.007 | SH3 and cysteine rich domain 3 |
| VKORC1 | 4.188 | 0.001 | 0.008 | Vitamin K epoxide reductase complex, subunit 1 |
| RNF32 | 4.171 | 0.001 | 0.008 | Ring finger protein 32 |
| FAM69A | 3.157 | 0.001 | 0.008 | Family with sequence similarity 69, member A |
| MFN2 | 4.574 | 0.001 | 0.008 | Mitofusin 2 |
| TNFRSF25 | 3.608 | 0.002 | 0.008 | Tumor necrosis factor receptor superfamily, member 25 |
| DCLRE1C | 2.249 | 0.002 | 0.008 | DNA cross-link repair 1C |
| G3BP1 | 5.532 | 0.002 | 0.008 | GTPase activating protein (SH3 domain) binding protein 1 |
| TSTD2 | 2.449 | 0.002 | 0.008 | Thiosulfate sulfurtransferase (rhodanese)-like domain containing 2 |
| PPARD | 2.386 | 0.002 | 0.009 | Peroxisome proliferator-activated receptor delta |
| E2F7 | 3.209 | 0.003 | 0.010 | E2F transcription factor 7 |
| LRRC1 | 3.610 | 0.003 | 0.010 | Leucine-rich repeat-containing 1 |
| PABPC4 | 4.588 | 0.003 | 0.011 | Poly(A) binding protein, cytoplasmic 4 (inducible form) |
| PAN3 | 2.918 | 0.003 | 0.011 | PAN3 poly(A) specific ribonuclease subunit |
| SLITRK6 | 2.372 | 0.003 | 0.011 | SLIT and NTRK-like family, member 6 |
| LRRC28 | 2.118 | 0.004 | 0.012 | Leucine-rich repeat-containing 28 |
| NFIA | 2.799 | 0.004 | 0.012 | Nuclear factor I/A |
| LIMD1 | 3.078 | 0.005 | 0.013 | LIM domains containing 1 |
| AHSA2 | 3.386 | 0.005 | 0.013 | AHA1, activator of heat shock 90kDa protein ATPase homolog 2 (yeast) |
| STK3 | 3.704 | 0.005 | 0.013 | Serine/threonine kinase 3 |
| AGER | 2.129 | 0.005 | 0.013 | Advanced glycosylation end-product-specific receptor |
| TMEM170B | 2.057 | 0.006 | 0.013 | Transmembrane protein 170B |
| PCM1 | 3.421 | 0.006 | 0.013 | Pericentriolar material 1 |
| KDELC2 | 2.870 | 0.006 | 0.013 | KDEL (Lys-Asp-Glu-Leu) containing 2 |
| ALDH1A3 | 2.978 | 0.006 | 0.013 | Aldehyde dehydrogenase 1 family, member A3 |
| MED20 | 2.466 | 0.008 | 0.015 | Mediator complex subunit 20 |
| SEPN1 | 2.196 | 0.008 | 0.016 | selenoprotein N, 1 |
| ARAP2 | 2.436 | 0.008 | 0.016 | ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 2 |
| ZNF550 | 2.510 | 0.009 | 0.016 | Zinc finger protein 550 |
| HIST2H2BF | 2.396 | 0.009 | 0.016 | Histone cluster 2, H2bf |
| SH3RF3 | 2.007 | 0.013 | 0.020 | SH3 domain-containing ring finger 3 |
| C3AR1 | 2.025 | 0.014 | 0.021 | Complement component 3a receptor 1 |
| ZNF808 | 2.568 | 0.014 | 0.021 | Zinc finger protein 808 |
| DOCK7 | 2.394 | 0.014 | 0.021 | Dedicator of cytokinesis 7 |
| PLCH1 | 2.144 | 0.014 | 0.021 | Phospholipase C, eta 1 |
| CLDN12 | 2.150 | 0.017 | 0.023 | Claudin 12 |
| POU5F1 | 2.168 | 0.018 | 0.025 | POU class 5 homeobox 1 |
| CEP170 | 2.189 | 0.032 | 0.036 | Centrosomal protein 170kDa |
| RAPGEF6 | 2.540 | 0.043 | 0.045 | Rap guanine nucleotide exchange factor (GEF) 6 |
| CLIP3 | 2.340 | 0.048 | 0.048 | CAP-GLY domain-containing linker protein 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, S.B.M.; Radwan, N.; Amer, S.; Saheb Sharif-Askari, N.; Mahdami, A.; Samara, K.A.; Halwani, R.; Jelinek, H.F. Assessing the Link between Diabetic Metabolic Dysregulation and Breast Cancer Progression. Int. J. Mol. Sci. 2023, 24, 11816. https://doi.org/10.3390/ijms241411816
Ahmed SBM, Radwan N, Amer S, Saheb Sharif-Askari N, Mahdami A, Samara KA, Halwani R, Jelinek HF. Assessing the Link between Diabetic Metabolic Dysregulation and Breast Cancer Progression. International Journal of Molecular Sciences. 2023; 24(14):11816. https://doi.org/10.3390/ijms241411816
Chicago/Turabian StyleAhmed, Samrein B. M., Nada Radwan, Sara Amer, Narjes Saheb Sharif-Askari, Amena Mahdami, Kamel A. Samara, Rabih Halwani, and Herbert F. Jelinek. 2023. "Assessing the Link between Diabetic Metabolic Dysregulation and Breast Cancer Progression" International Journal of Molecular Sciences 24, no. 14: 11816. https://doi.org/10.3390/ijms241411816
APA StyleAhmed, S. B. M., Radwan, N., Amer, S., Saheb Sharif-Askari, N., Mahdami, A., Samara, K. A., Halwani, R., & Jelinek, H. F. (2023). Assessing the Link between Diabetic Metabolic Dysregulation and Breast Cancer Progression. International Journal of Molecular Sciences, 24(14), 11816. https://doi.org/10.3390/ijms241411816