
DeepSATA: A Deep Learning-Based Sequence Analyzer Incorporating the Transcription Factor Binding Affinity to Dissect the Effects of Non-Coding Genetic Variants

Abstract
1. Introduction
2. Results
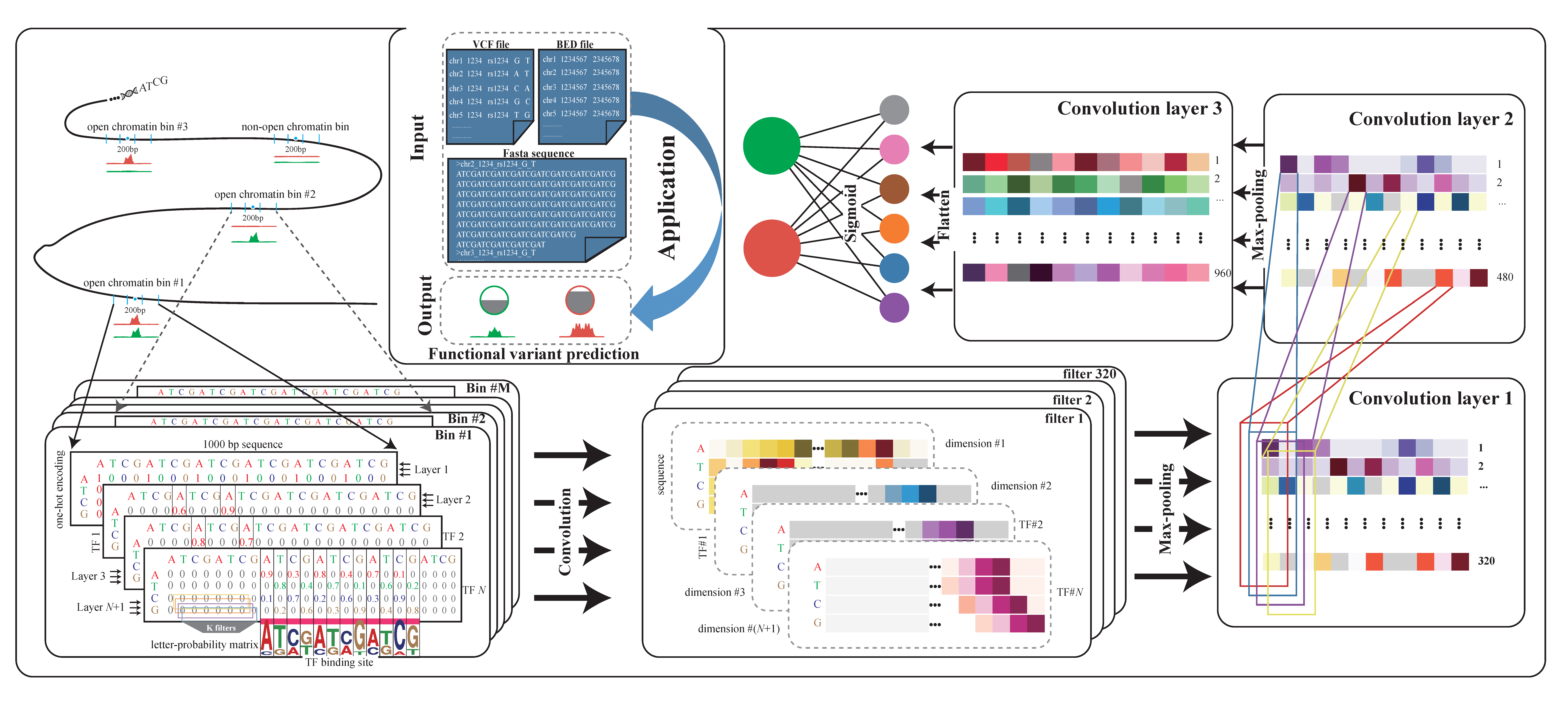
2.1. Overview of DeepSATA
2.2. DeepSATA Consistently Outperformed DeepSEA and Basset in AUC Results
2.3. DeepSATA Achieved Reliable Cross-Species Predictions
2.4. DeepSATA Achieved Significant Accuracy for Biological Functions
2.5. Case Study of a Regulatory SNP Identified by DeepSATA
2.6. DeepSATA-Improved Phenotype Prediction Performance
3. Discussion
4. Materials and Methods
4.1. Data Collection for Constructing the Model
4.2. Model Training in DeepSATA
4.3. Cross-Species Predictions
4.4. Predicting Impacts of Variants on Open Chromatin
4.5. Enrichment Analysis on SNPs That Are Significantly Impacting Open Chromatin
4.6. Phenotype Predictions Using rrBLUP
4.7. Sequence Similarity Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association studies |
| DL | Deep learning |
| DNA | Deoxyribonucleic acid |
| TF | Transcription factor |
| DNN | Deep neural network |
| CNN | Convolutional neural network |
| ROC | Receiver operating characteristic curve |
| AUC | Area under the curve |
| bp | Base pairs |
| OCB | Open chromatin bin |
| OCR | Open chromatin region |
| SNP | Single-nucleotide polymorphism |
| FDR | False discovery rate |
| PCC | Pearson correlation coefficients |
References
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Harley, I.T.W.; Lu, X.; Zhou, T.; Xu, N.; Yao, C.; Qin, Y.; Ouyang, Y.; Ma, J.; Zhu, X.; et al. SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression. Nat. Commun. 2021, 12, 135. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Mok, K.Y.; Kwok, T.C.Y.; Mok, V.C.T.; Guo, Q.; Ip, F.C.; Chen, Y.; Mullapudi, N.; Alzheimer’s Disease Neuroimaging Initiative; et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat. Commun. 2019, 10, 3310. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, Y.; Yang, Y.; Yi, G.; Lian, J.; Xie, B.; Yao, Y.; Chen, M.; Niu, Y.; Liu, L.; et al. Integration of multi-omics data reveals cis-regulatory variants that are associated with phenotypic differentiation of eastern from western pigs. Genet. Sel. Evol. 2022, 54, 62. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Yao, Y.; Yin, H.; Cai, Z.; Wang, Y.; Bai, L.; Kern, C.; Halstead, M.; Chanthavixay, G.; Trakooljul, N.; et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat. Commun. 2021, 12, 5848. [Google Scholar] [CrossRef]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef]
- Maher, B. ENCODE: The human encyclopaedia. Nature 2012, 489, 46–48. [Google Scholar] [CrossRef]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Giuffra, E.; Tuggle, C.K.; Consortium, F. Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2019, 7, 65–88. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, G.; Avsec, Z.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016, 26, 990–999. [Google Scholar] [CrossRef]
- Trieu, T.; Martinez-Fundichely, A.; Khurana, E. DeepMILO: A deep learning approach to predict the impact of non-coding sequence variants on 3D chromatin structure. Genome Biol. 2020, 21, 79. [Google Scholar] [CrossRef]
- Pei, G.; Hu, R.; Jia, P.; Zhao, Z. DeepFun: A deep learning sequence-based model to decipher non-coding variant effect in a tissue- and cell type-specific manner. Nucleic Acids Res. 2021, 49, W131–W139. [Google Scholar] [CrossRef]
- Kelley, D.R. Cross-species regulatory sequence activity prediction. PLoS Comput. Biol. 2020, 16, e1008050. [Google Scholar] [CrossRef]
- Chen, L.; Fish, A.E.; Capra, J.A. Prediction of gene regulatory enhancers across species reveals evolutionarily conserved sequence properties. PLoS Comput. Biol. 2018, 14, e1006484. [Google Scholar] [CrossRef]
- Villar, D.; Flicek, P.; Odom, D.T. Evolution of transcription factor binding in metazoans—Mechanisms and functional implications. Nat. Rev. Genet. 2014, 15, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Perez, N.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Z.; Bruce, H.; Kemp, R.A.; Charagu, P.; Miar, Y.; Yang, T.; Plastow, G. Genome-wide association studies (GWAS) identify a QTL close to PRKAG3 affecting meat pH and colour in crossbred commercial pigs. BMC Genet. 2015, 16, 33. [Google Scholar] [CrossRef]
- Szulc, K.; Wojtysiak, D.; Migdał, Ł.; Migdał, W. The Muscle Fibre Characteristics and the Meat Quality of m. longissimus thoracis from Polish Native Złotnicka Spotted Pigs and the Crossbreed Fatteners from the Crossing of Duroc and Polish Large White Boars. Appl. Sci. 2022, 12, 3051. [Google Scholar] [CrossRef]
- Liu, Q.; Long, Y.; Zhang, Y.F.; Zhang, Z.Y.; Yang, B.; Chen, C.Y.; Huang, L.S.; Su, Y. Phenotypic and genetic correlations of pork myoglobin content with meat colour and other traits in an eight breed-crossed heterogeneous population. Animal 2021, 15, 100364. [Google Scholar] [CrossRef]
- Oliveira, H.C.; Derks, M.F.L.; Lopes, M.S.; Madsen, O.; Harlizius, B.; van Son, M.; Grindflek, E.H.; Godia, M.; Gjuvsland, A.B.; Otto, P.I.; et al. Fine Mapping of a Major Backfat QTL Reveals a Causal Regulatory Variant Affecting the CCND2 Gene. Front. Genet. 2022, 13, 871516. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Yang, R.; Guo, X.; Zhu, D.; Tan, C.; Bian, C.; Ren, J.; Huang, Z.; Zhao, Y.; Cai, G.; Liu, D.; et al. Accelerated deciphering of the genetic architecture of agricultural economic traits in pigs using a low-coverage whole-genome sequencing strategy. Gigascience 2021, 10, giab048. [Google Scholar] [CrossRef]
- Chen, C.; Hou, J.; Shi, X.; Yang, H.; Birchler, J.A.; Cheng, J. DeepGRN: Prediction of transcription factor binding site across cell-types using attention-based deep neural networks. BMC Bioinform. 2021, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hua, K.; Zhang, X.; Wong, W.H.; Jiang, R. DeepCAGE: Incorporating transcription factors in genome-wide predic-tion of chromatin accessibility. Genom. Proteom. Bioinform. 2022, 20, 496–507. [Google Scholar] [CrossRef]
- ENCODE. Available online: https://www.encodeproject.org/ (accessed on 5 May 2023).
- Zhao, Y.; Hou, Y.; Xu, Y.; Luan, Y.; Zhou, H.; Qi, X.; Hu, M.; Wang, D.; Wang, Z.; Fu, Y.; et al. A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome. Nat. Commun. 2021, 12, 2217. [Google Scholar] [CrossRef]
- Kern, C.; Wang, Y.; Xu, X.; Pan, Z.; Halstead, M.; Chanthavixay, G.; Saelao, P.; Waters, S.; Xiang, R.; Chamberlain, A.; et al. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 2021, 12, 1821. [Google Scholar] [CrossRef]
- Li, Q.; Brown, J.B.; Huang, H.; Bickel, P.J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011, 5, 1752–1779. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- PigQTLdb. Available online: https://www.animalgenome.org/cgi-bin/QTLdb/SS/index (accessed on 15 May 2021).
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- GigaDB. Available online: http://gigadb.org/dataset/100894 (accessed on 15 May 2023).
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- LiftOver. Available online: http://genome.ucsc.edu/cgi-bin/hgLiftOver (accessed on 11 August 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Tissue Number | Breed Number | Gender Number | Sample Number | Mean OCR Number a | JASPAR2022 Motif |
|---|---|---|---|---|---|---|
| Mus musculus | 18 | 0 | 2 | 32 | 36,727 | Mus musculus |
| Sus scrofa | 5 | 4 | 0 | 16 | 124,190 | Vertebrata |
| Bos taurus | 8 | 0 | 0 | 8 | 84,463 | Vertebrata |
| Homo sapiens | 35 | 0 | 0 | 35 | 168,702 | Homo sapiens |
| Gallus gallus | 8 | 0 | 0 | 8 | 25,362 | Vertebrata |
| Bins a | Average Coverage b | TF.Ratio c | DeepSATA | DeepSEA | |
|---|---|---|---|---|---|
| mice | 1,081,545 | 170.61 | 0.162 | 0.8536 | 0.7962 |
| pigs | 1,586,677 | 165.84 | 0.0946 | 0.7785 | 0.7746 |
| cattle | 1,872,585 | 167.14 | 0.0768 | 0.7721 | 0.7686 |
| human | 3,337,706 | 156.86 | 0.0763 | 0.7592 | 0.7554 |
| chickens | 570,905 | 166.76 | 0.0868 | 0.7444 | 0.7358 |
| Bins a | Average Coverage b | TF.Ratio c | DeepSATA | DeepSEA | |
|---|---|---|---|---|---|
| Duroc | 884,195 | 167.08 | 0.01178 | 0.7891 | 0.7863 |
| ES | 1,307,825 | 163.45 | 0.0122 | 0.7725 | 0.7693 |
| LW | 748,865 | 167.86 | 0.0120 | 0.782 | 0.7766 |
| MS | 779,553 | 161.46 | 0.0120 | 0.7674 | 0.762 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Fu, Y.; Bao, Y.; Wang, Z.; Lei, B.; Zheng, W.; Wang, C.; Liu, Y. DeepSATA: A Deep Learning-Based Sequence Analyzer Incorporating the Transcription Factor Binding Affinity to Dissect the Effects of Non-Coding Genetic Variants. Int. J. Mol. Sci. 2023, 24, 12023. https://doi.org/10.3390/ijms241512023
Ma W, Fu Y, Bao Y, Wang Z, Lei B, Zheng W, Wang C, Liu Y. DeepSATA: A Deep Learning-Based Sequence Analyzer Incorporating the Transcription Factor Binding Affinity to Dissect the Effects of Non-Coding Genetic Variants. International Journal of Molecular Sciences. 2023; 24(15):12023. https://doi.org/10.3390/ijms241512023
Chicago/Turabian StyleMa, Wenlong, Yang Fu, Yongzhou Bao, Zhen Wang, Bowen Lei, Weigang Zheng, Chao Wang, and Yuwen Liu. 2023. "DeepSATA: A Deep Learning-Based Sequence Analyzer Incorporating the Transcription Factor Binding Affinity to Dissect the Effects of Non-Coding Genetic Variants" International Journal of Molecular Sciences 24, no. 15: 12023. https://doi.org/10.3390/ijms241512023
APA StyleMa, W., Fu, Y., Bao, Y., Wang, Z., Lei, B., Zheng, W., Wang, C., & Liu, Y. (2023). DeepSATA: A Deep Learning-Based Sequence Analyzer Incorporating the Transcription Factor Binding Affinity to Dissect the Effects of Non-Coding Genetic Variants. International Journal of Molecular Sciences, 24(15), 12023. https://doi.org/10.3390/ijms241512023