
Genome-Wide Profiling of Endogenous Single-Stranded DNA Using the SSiNGLe-P1 Method

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Concept of SSiNGLe-P1: A P1 Endonuclease-Based Strategy for the Detection of essDNA Regions
2.2. Application of SSiNGLe-P1 to Evaluate the Strand-Displacement Model of mtDNA Replication
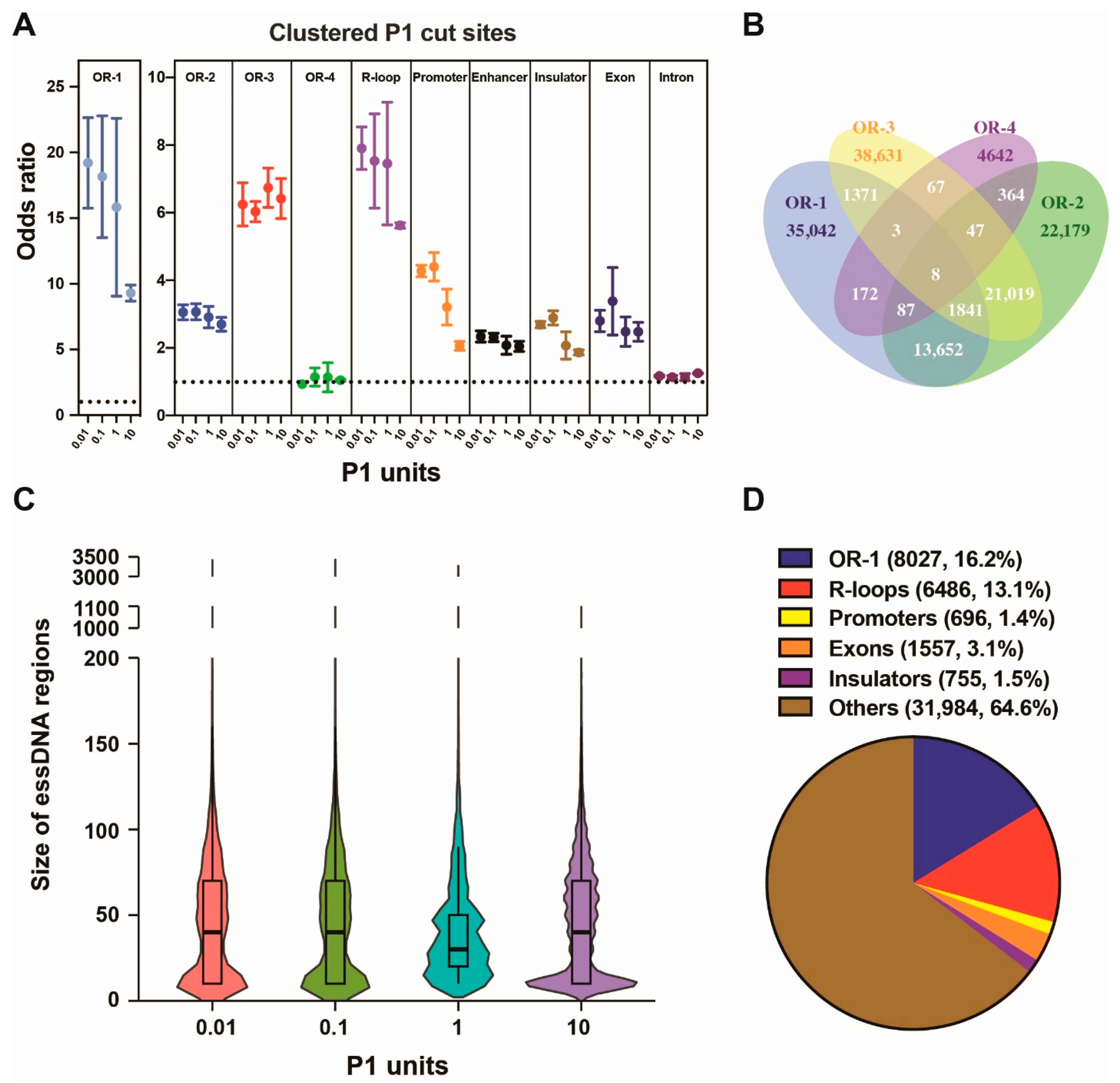
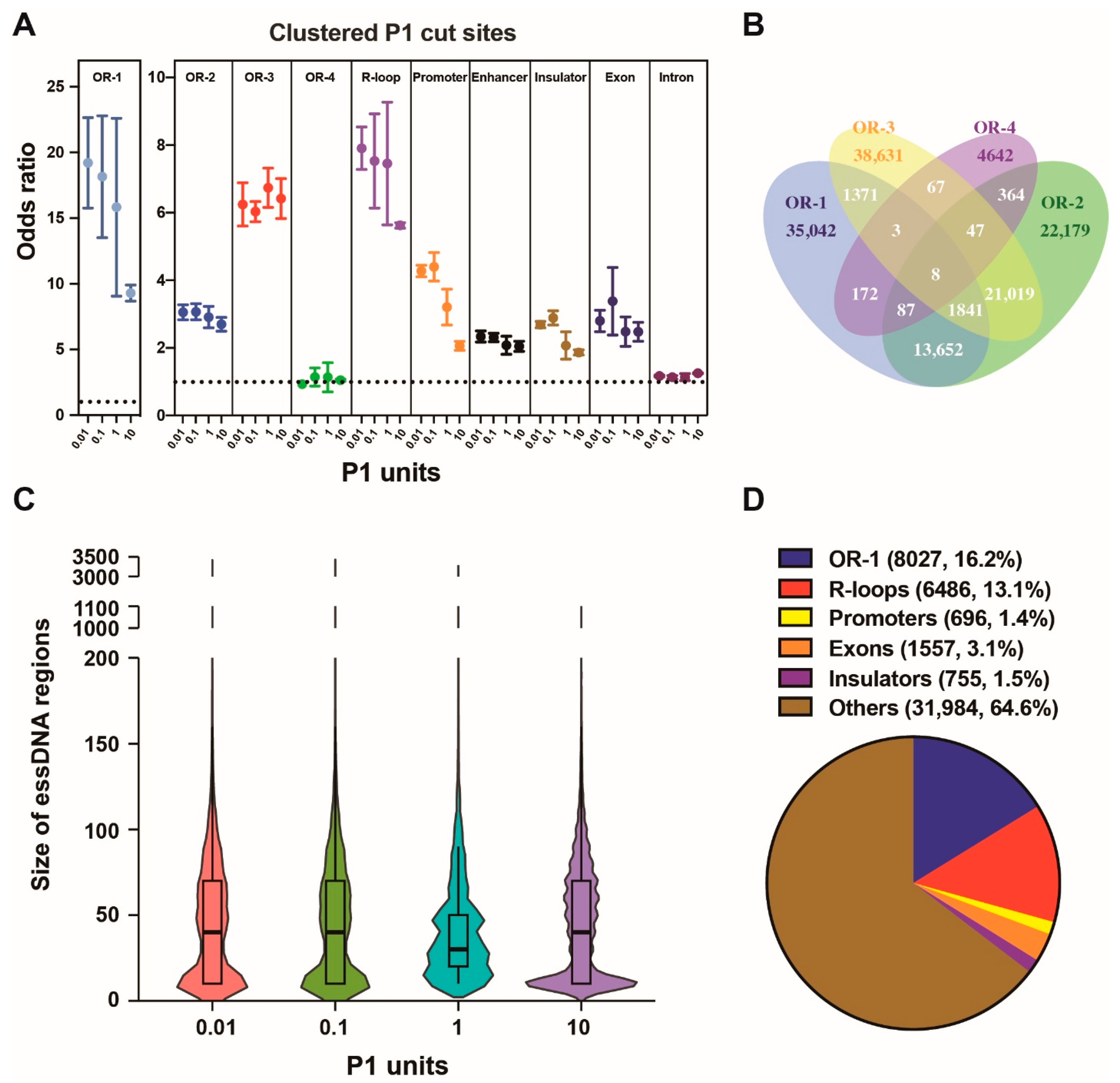
2.3. Detection of essDNA Regions in the Nuclear Genome
2.4. Properties of the Nuclear essDNA Regions Identified by SSiNGLe-P1
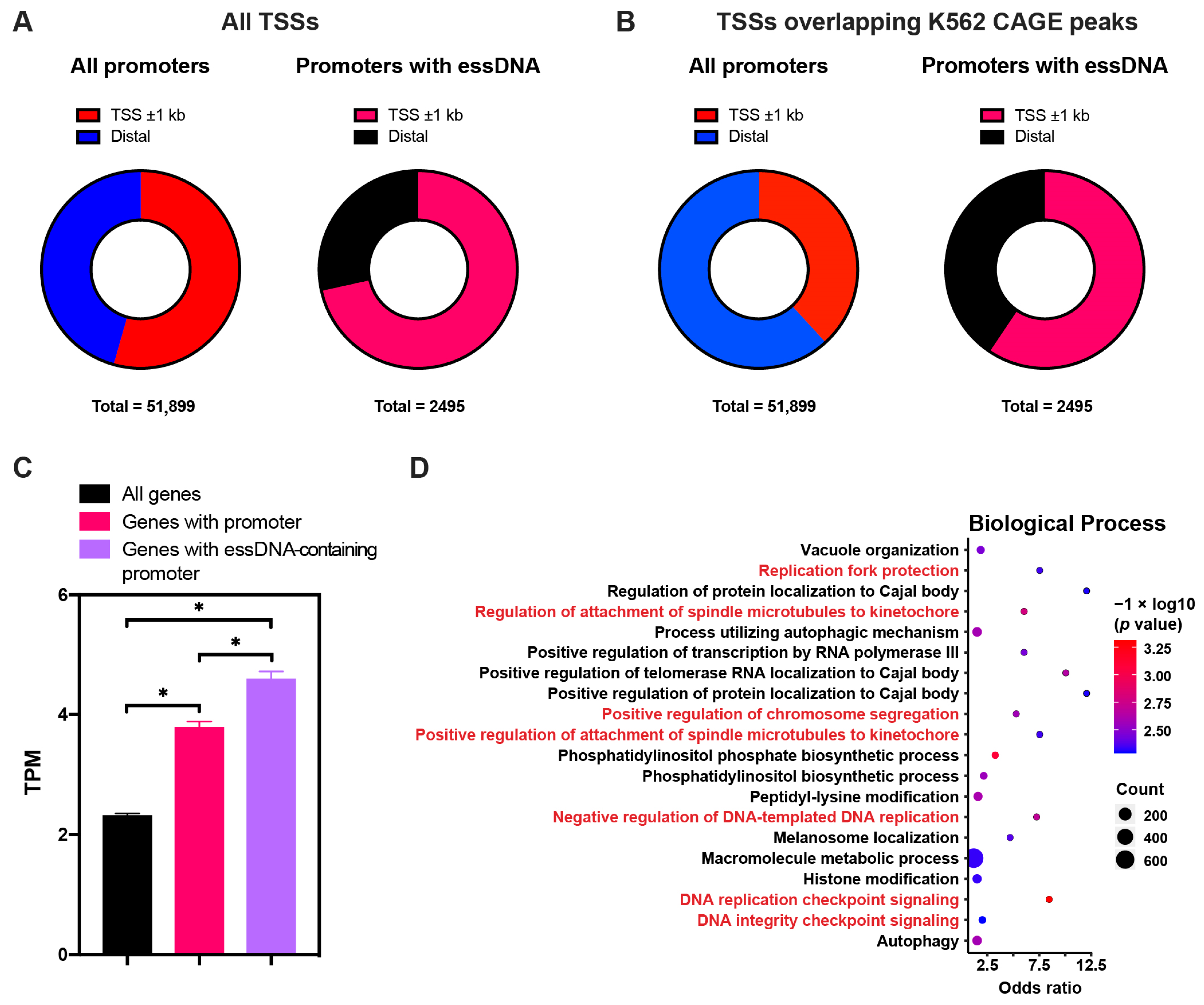
2.5. The essDNA Regions Tend to Occur in Promoters of Highly Expressed Annotated Genes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. SSiNGLe-P1: Wet Lab
4.3. SSiNGLe-P1: Bioinformatics
4.4. Simulation Analysis
4.5. RNA-Seq Analysis
4.6. General Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicholls, T.J.; Minczuk, M. In D-Loop: 40 Years of Mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomeres Do D-Loop-T-Loop. Cell 1999, 97, 419–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Collingwood, D.; Boeck, M.E.; Fox, L.A.; Alvino, G.M.; Fangman, W.L.; Raghuraman, M.K.; Brewer, B.J. Genomic Mapping of Single-Stranded DNA in Hydroxyurea-Challenged Yeasts Identifies Origins of Replication. Nat. Cell Biol. 2006, 8, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Holt, I.J.; Reyes, A. Human Mitochondrial DNA Replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a012971. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Lyu, R.; You, Q.; He, C. Kethoxal-Assisted Single-Stranded DNA Sequencing Captures Global Transcription Dynamics and Enhancer Activity in Situ. Nat. Methods 2020, 17, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Wojtowicz, D.; Yamane, A.; Resch, W.; Kieffer-Kwon, K.-R.; Bandle, R.; Nelson, S.; Nakahashi, H.; Awasthi, P.; Feigenbaum, L.; et al. Global Regulation of Promoter Melting in Naive Lymphocytes. Cell 2013, 153, 988–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantele, S.C.S.; Lisby, M.; Pfander, B. Quantitative Sensing and Signalling of Single-Stranded DNA during the DNA Damage Response. Nat. Commun. 2019, 10, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [Green Version]
- Hisano, O.; Ito, T.; Miura, F. Short Single-Stranded DNAs with Putative Non-Canonical Structures Comprise a New Class of Plasma Cell-Free DNA. BMC Biol. 2021, 19, 225. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Wojtowicz, D.; Baranello, L.; Yamane, A.; Nelson, S.; Resch, W.; Kieffer-Kwon, K.-R.; Benham, C.J.; Casellas, R.; Przytycka, T.M.; et al. Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst. 2017, 4, 344–356.e7. [Google Scholar] [CrossRef] [Green Version]
- Pham, P.; Shao, Y.; Cox, M.M.; Goodman, M.F. Genomic Landscape of Single-Stranded DNA Gapped Intermediates in Escherichia Coli. Nucleic Acids Res. 2022, 50, 937–951. [Google Scholar] [CrossRef]
- Pham, P.; Wood, E.A.; Cox, M.M.; Goodman, M.F. RecA and SSB Genome-Wide Distribution in SsDNA Gaps and Ends in Escherichia Coli. Nucleic Acids Res. 2023, 51, 5527–5546. [Google Scholar] [CrossRef]
- MacDonald, G.H.; Itoh-Lindstrom, Y.; Ting, J.P. The Transcriptional Regulatory Protein, YB-1, Promotes Single-Stranded Regions in the DRA Promoter. J. Biol. Chem. 1995, 270, 3527–3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Pedigo, N.; Shenoy, S.; Khalili, K.; Kaetzel, D.M. Puralpha Activates PDGF-A Gene Transcription via Interactions with a G-Rich, Single-Stranded Region of the Promoter. Gene 2005, 348, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Jiang, J.; Ren, Y.; Zhao, Z. The Single-Stranded DNA-Binding Protein WHIRLY1 Represses WRKY53 Expression and Delays Leaf Senescence in a Developmental Stage-Dependent Manner in Arabidopsis. Plant Physiol. 2013, 163, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R Loops: New Modulators of Genome Dynamics and Function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chédin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, D.F.; Wang, G.G. R-Loops: Formation, Function, and Relevance to Cell Stress. Cell Stress 2019, 3, 38–46. [Google Scholar] [CrossRef] [Green Version]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA Polymerase II Pausing Mitigates BRCA1-Associated R-Loop Accumulation and Tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, C.; Griffith, E.; Brady, G.; Lydall, D. Quantitative Amplification of Single-Stranded DNA (QAOS) Demonstrates That Cdc13-1 Mutants Generate SsDNA in a Telomere to Centromere Direction. Nucleic Acids Res. 2001, 29, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Holstein, E.-M.; Lydall, D. Quantitative Amplification of Single-Stranded DNA. Methods Mol. Biol. 2012, 920, 323–339. [Google Scholar] [CrossRef]
- Kilgas, S.; Singh, A.N.; Paillas, S.; Then, C.-K.; Torrecilla, I.; Nicholson, J.; Browning, L.; Vendrell, I.; Konietzny, R.; Kessler, B.M.; et al. P97/VCP Inhibition Causes Excessive MRE11-Dependent DNA End Resection Promoting Cell Killing after Ionizing Radiation. Cell Rep. 2021, 35, 109153. [Google Scholar] [CrossRef]
- Kilgas, S.; Kiltie, A.E.; Ramadan, K. Immunofluorescence Microscopy-Based Detection of SsDNA Foci by BrdU in Mammalian Cells. STAR Protoc. 2021, 2, 100978. [Google Scholar] [CrossRef]
- Raderschall, E.; Golub, E.I.; Haaf, T. Nuclear Foci of Mammalian Recombination Proteins Are Located at Single-Stranded DNA Regions Formed after DNA Damage. Proc. Natl. Acad. Sci. USA 1999, 96, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Chatzidoukaki, O.; Stratigi, K.; Goulielmaki, E.; Niotis, G.; Akalestou-Clocher, A.; Gkirtzimanaki, K.; Zafeiropoulos, A.; Altmüller, J.; Topalis, P.; Garinis, G.A. R-Loops Trigger the Release of Cytoplasmic SsDNAs Leading to Chronic Inflammation upon DNA Damage. Sci. Adv. 2021, 7, eabj5769. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Kono, N.; Tomita, M.; Arakawa, K. Strand-Specific Single-Stranded DNA Sequencing (4S-Seq) of E. Coli Genomes. Bio. Protoc. 2019, 9, e3329. [Google Scholar] [CrossRef] [PubMed]
- Buhler, C.; Borde, V.; Lichten, M. Mapping Meiotic Single-Strand DNA Reveals a New Landscape of DNA Double-Strand Breaks in Saccharomyces Cerevisiae. PLoS Biol. 2007, 5, e324. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Salazar-García, L.; Gao, F.; Wahlestedt, T.; Wu, C.-L.; Han, X.; Cai, Y.; Xu, D.; Wang, F.; Tang, L.; et al. Novel Approach Reveals Genomic Landscapes of Single-Strand DNA Breaks with Nucleotide Resolution in Human Cells. Nat. Commun. 2019, 10, 5799. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Kuninaka, A.; Yoshino, H. Purification of a Nuclease from Penicillium Citrinum. Agric. Biol. Chem. 1974, 38, 777–783. [Google Scholar] [CrossRef]
- Volbeda, A.; Lahm, A.; Sakiyama, F.; Suck, D. Crystal Structure of Penicillium Citrinum P1 Nuclease at 2.8 A Resolution. EMBO J. 1991, 10, 1607–1618. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-Strand Break Repair and Genetic Disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T. Days Weaving the Lagging Strand Synthesis of DNA—A Personal Recollection of the Discovery of Okazaki Fragments and Studies on Discontinuous Replication Mechanism. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 322–338. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Kuninaka, A.; Yoshino, H. Substrate Specificity of Nuclease P1. Agric. Biol. Chem. 1974, 38, 1555–1561. [Google Scholar] [CrossRef]
- Falkenberg, M. Mitochondrial DNA Replication in Mammalian Cells: Overview of the Pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar] [CrossRef]
- Holt, I.J.; Lorimer, H.E.; Jacobs, H.T. Coupled Leading- and Lagging-Strand Synthesis of Mammalian Mitochondrial DNA. Cell 2000, 100, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Bowmaker, M.; Yang, M.Y.; Yasukawa, T.; Reyes, A.; Jacobs, H.T.; Huberman, J.A.; Holt, I.J. Mammalian Mitochondrial DNA Replicates Bidirectionally from an Initiation Zone. J. Biol. Chem. 2003, 278, 50961–50969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasukawa, T.; Reyes, A.; Cluett, T.J.; Yang, M.-Y.; Bowmaker, M.; Jacobs, H.T.; Holt, I.J. Replication of Vertebrate Mitochondrial DNA Entails Transient Ribonucleotide Incorporation throughout the Lagging Strand. EMBO J. 2006, 25, 5358–5371. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Kazak, L.; Wood, S.R.; Yasukawa, T.; Jacobs, H.T.; Holt, I.J. Mitochondrial DNA Replication Proceeds via a “bootlace” Mechanism Involving the Incorporation of Processed Transcripts. Nucleic Acids Res. 2013, 41, 5837–5850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A Clustering Approach for Identification of Enriched Domains from Histone Modification ChIP-Seq Data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Miotto, B.; Ji, Z.; Struhl, K. Selectivity of ORC Binding Sites and the Relation to Replication Timing, Fragile Sites, and Deletions in Cancers. Proc. Natl. Acad. Sci. USA 2016, 113, E4810-9. [Google Scholar] [CrossRef]
- Picard, F.; Cadoret, J.-C.; Audit, B.; Arneodo, A.; Alberti, A.; Battail, C.; Duret, L.; Prioleau, M.-N. The Spatiotemporal Program of DNA Replication Is Associated with Specific Combinations of Chromatin Marks in Human Cells. PLoS Genet. 2014, 10, e1004282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.M.; Ryan, M.; Kim, R.; Zakas, A.L.; Fu, H.; Lin, C.M.; Reinhold, W.C.; Davis, S.R.; Bilke, S.; Liu, H.; et al. Genome-Wide Depletion of Replication Initiation Events in Highly Transcribed Regions. Genome Res. 2011, 21, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing Newly Replicated DNA Reveals Widespread Plasticity in Human Replication Timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef]
- Fiedler, U.; Timmers, H.T. Analysis of the Open Region of RNA Polymerase II Transcription Complexes in the Early Phase of Elongation. Nucleic Acids Res. 2001, 29, 2706–2714. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Discovery and Characterization of Chromatin States for Systematic Annotation of the Human Genome. Nat. Biotechnol. 2010, 28, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and Analysis of Chromatin State Dynamics in Nine Human Cell Types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [Green Version]
- The FANTOM Consortium and the RIKEN PMI and CLST (DGT). A Promoter-Level Mammalian Expression Atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, I.J. The Mitochondrial R-Loop. Nucleic Acids Res. 2019, 47, 5480–5489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Zhang, Y.; Cai, Y.; Tang, L.; Gao, F.; Xu, D.; Kapranov, P. Hotspots of Single-Strand DNA “Breakome” Are Enriched at Transcriptional Start Sites of Genes. Front. Mol. Biosci. 2022, 9, 895795. [Google Scholar] [CrossRef]
- Gordon, A. FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/download.html (accessed on 6 September 2018).
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.; Kent, W.J.; Clawson, H.; Kuhn, R.M.; Diekhans, M.; Haussler, D. The UCSC Known Genes. Bioinformatics 2006, 22, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, S.; Gentleman, R. Using GOstats to Test Gene Lists for GO Term Association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.; Huang, Y.; Luo, L.; Tang, L.; Lu, M.; Cao, H.; Wang, F.; Diao, Y.; Lyubchenko, L.; Kapranov, P. Genome-Wide Profiling of Endogenous Single-Stranded DNA Using the SSiNGLe-P1 Method. Int. J. Mol. Sci. 2023, 24, 12062. https://doi.org/10.3390/ijms241512062
Xu D, Huang Y, Luo L, Tang L, Lu M, Cao H, Wang F, Diao Y, Lyubchenko L, Kapranov P. Genome-Wide Profiling of Endogenous Single-Stranded DNA Using the SSiNGLe-P1 Method. International Journal of Molecular Sciences. 2023; 24(15):12062. https://doi.org/10.3390/ijms241512062
Chicago/Turabian StyleXu, Dongyang, Yu Huang, Lingcong Luo, Lu Tang, Meng Lu, Huifen Cao, Fang Wang, Yong Diao, Liudmila Lyubchenko, and Philipp Kapranov. 2023. "Genome-Wide Profiling of Endogenous Single-Stranded DNA Using the SSiNGLe-P1 Method" International Journal of Molecular Sciences 24, no. 15: 12062. https://doi.org/10.3390/ijms241512062
APA StyleXu, D., Huang, Y., Luo, L., Tang, L., Lu, M., Cao, H., Wang, F., Diao, Y., Lyubchenko, L., & Kapranov, P. (2023). Genome-Wide Profiling of Endogenous Single-Stranded DNA Using the SSiNGLe-P1 Method. International Journal of Molecular Sciences, 24(15), 12062. https://doi.org/10.3390/ijms241512062