

Glucocorticoid Treatment in Acute Respiratory Distress Syndrome: An Overview on Mechanistic Insights and Clinical Benefit
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Glucocorticoids
2.1. Endogenous GCs
2.1.1. GC Definition, Source, and Production
2.1.2. GCs Structure
2.1.3. GCs Mechanisms of Action
2.2. Exogenous GCs
2.2.1. Short-Acting GCs
2.2.2. Medium-Acting GCs
2.2.3. Long-Acting GCs
3. GCs Mechanism in SAP-SIRS-ALI Treatment
3.1. Core Pathology of SAP-SIRS-ALI
3.2. Research Progress of GC in SAP-SIRS-ALI
3.2.1. Glucocorticoid Receptor (GR)
3.2.2. Anti-Inflammatory Role of GCs
Suppression of Pro-Inflammatory Gene Expression by GCs
Regulation of Signal Transduction by GCs
Novel Anti-Inflammatory Mechanisms
Other Anti-Inflammatory Pathways Regulated by GCs
3.2.3. Microcirculation Improvement by GCs
3.2.4. The Protective Effect of GCs on Lung Tissue
3.2.5. Immunosuppressive Effects
3.2.6. Endothelial Cell Protection
4. Clinical Use of GC in SAP-ALI/ARDS Treatment
4.1. Clinical Treatment for SAP-ALI/ARDS
4.2. The Application Status of GCs
4.3. Administration of GCs
4.4. Limitation of GCs
4.4.1. AP Induction by GCs
4.4.2. Infection Caused by GCs
4.4.3. Hemorrhage Caused by GCs
4.4.4. Other Effects of GCs
5. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kemppainen, R.J.; Behrend, E.N. Adrenal physiology. Vet. Clin. N. Am. Small Anim. Pract. 1997, 27, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Shimba, A.; Ikuta, K. Glucocorticoids Regulate Circadian Rhythm of Innate and Adaptive Immunity. Front. Immunol. 2020, 11, 2143. [Google Scholar] [CrossRef] [PubMed]
- Wilcke, J.R.; Davis, L.E. Review of glucocorticoid pharmacology. Vet. Clin. N. Am. Small Anim. Pract. 1982, 12, 3–17. [Google Scholar] [CrossRef]
- Meduri, G.U.; Annane, D.; Confalonieri, M.; Chrousos, G.P.; Rochwerg, B.; Busby, A.; Ruaro, B.; Meibohm, B. Pharmacological principles guiding prolonged glucocorticoid treatment in ARDS. Intensive Care Med. 2020, 46, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Ferrando, C.; Martínez, D.; Ambrós, A.; Muñoz, T.; Soler, J.A.; Aguilar, G.; Alba, F.; González-Higueras, E.; Conesa, L.A.; et al. Dexamethasone treatment for the acute respiratory distress syndrome: A multicentre, randomised controlled trial. Lancet Respir. Med. 2020, 8, 267–276. [Google Scholar] [CrossRef]
- Dubashynskaya, N.V.; Bokatyi, A.N.; Skorik, Y.A. Dexamethasone Conjugates: Synthetic Approaches and Medical Prospects. Biomedicines 2021, 9, 341. [Google Scholar] [CrossRef]
- Salton, F.; Confalonieri, P.; Meduri, G.U.; Santus, P.; Harari, S.; Scala, R.; Lanini, S.; Vertui, V.; Oggionni, T.; Caminati, A.; et al. Prolonged Low-Dose Methylprednisolone in Patients with Severe COVID-19 Pneumonia. Open Forum Infect. Dis. 2020, 7, ofaa421. [Google Scholar] [CrossRef]
- Xuan, N.; Zhang, X.; Hu, W.; Chen, G.; Wang, Y.; Zhang, S.; Cui, W.; Zhang, G. Effects of the working experience, educational background, professional titles, and hospital grades of intensive care unit doctors on clinical glucocorticoid use in acute respiratory distress syndrome. Medicine 2022, 101, e29021. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [Green Version]
- Behrend, E.N.; Kemppainen, R.J. Glucocorticoid therapy. Pharmacology, indications, and complications. Vet. Clin. N. Am. Small Anim. Pract. 1997, 27, 187–213. [Google Scholar] [CrossRef]
- Spiga, F.; Walker, J.J.; Terry, J.R.; Lightman, S.L. HPA axis-rhythms. Compr. Physiol. 2014, 4, 1273–1298. [Google Scholar] [PubMed]
- Huang, J.; Jia, R.; Brunner, T. Local synthesis of immunosuppressive glucocorticoids in the intestinal epithelium regulates anti-viral immune responses. Cell. Immunol. 2018, 334, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jozic, I.; Stojadinovic, O.; Kirsner, R.S.; Tomic-Canic, M. Stressing the steroids in skin: Paradox or fine-tuning. J. Investig. Dermatol. 2014, 134, 2869–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noti, M.; Sidler, D.; Brunner, T. Extra-adrenal glucocorticoid synthesis in the intestinal epithelium: More than a drop in the ocean. Semin. Immunopathol. 2009, 31, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Talaber, G.; Jondal, M.; Okret, S. Local glucocorticoid production in the thymus. Steroids 2015, 103, 58–63. [Google Scholar] [CrossRef]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24. [Google Scholar] [CrossRef] [Green Version]
- Upton, G.V.; Amatruda, T.T., Jr. Evidence for the presence of tumor peptides with corticotropin-releasing-factor-like activity in the ectopic ACTH syndrome. N. Engl. J. Med. 1971, 285, 419–424. [Google Scholar] [CrossRef]
- Miller, W.L. Androgen synthesis in adrenarche. Rev. Endocr. Metab. Disord. 2009, 10, 3–17. [Google Scholar] [CrossRef]
- Buchwald, P.; Bodor, N. Soft glucocorticoid design: Structural elements and physicochemical parameters determining receptor-binding affinity. Pharmazie 2004, 59, 396–404. [Google Scholar]
- Donatti, T.L.; Koch, V.H.; Takayama, L.; Pereira, R.M. Effects of glucocorticoids on growth and bone mineralization. J. Pediatr. 2011, 87, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Vegiopoulos, A.; Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol. 2007, 275, 43–61. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, U.A.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.M.; Gomez-Sanchez, C.E. The ubiquitous mineralocorticoid receptor: Clinical implications. Curr. Hypertens. Rep. 2012, 14, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Joëls, M. Impact of glucocorticoids on brain function: Relevance for mood disorders. Psychoneuroendocrinology 2011, 36, 406–414. [Google Scholar] [CrossRef]
- Tatomir, A.; Micu, C.; Crivii, C. The impact of stress and glucocorticoids on memory. Clujul Med. 2014, 87, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [Green Version]
- De Bosscher, K.; Haegeman, G. Minireview: Latest perspectives on antiinflammatory actions of glucocorticoids. Mol. Endocrinol. 2009, 23, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.A. Historical overview of nuclear receptors. J. Steroid Biochem. Mol. Biol. 2016, 157, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.L. Plasma steroid-binding proteins: Primary gatekeepers of steroid hormone action. J. Endocrinol. 2016, 230, R13–R25. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.L.; Smith, C.L.; Paterson, N.A.; Sibbald, W.J. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J. Clin. Endocrinol. Metab. 1990, 71, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Draper, N.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J. Endocrinol. 2005, 186, 251–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truss, M.; Beato, M. Steroid hormone receptors: Interaction with deoxyribonucleic acid and transcription factors. Endocr. Rev. 1993, 14, 459–479. [Google Scholar] [PubMed]
- Yeager, M.P.; Pioli, P.A.; Guyre, P.M. Cortisol exerts bi-phasic regulation of inflammation in humans. Dose-Response 2011, 9, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.Q.; Lu, J.X.; Deng, Y.J. Glucocorticoids offer protection against myocardial injury in a murine model of sepsis. Int. J. Clin. Exp. Med. 2015, 8, 12211–12218. [Google Scholar] [PubMed]
- Choudhury, S.; Tan, T.; Lazarus, K.; Meeran, K. The use of prednisolone versus dual-release hydrocortisone in the treatment of hypoadrenalism. Endocr. Connect. 2021, 10, R66–R76. [Google Scholar] [CrossRef]
- Frey, B.M.; Frey, F.J. Clinical pharmacokinetics of prednisone and prednisolone. Clin. Pharmacokinet. 1990, 19, 126–146. [Google Scholar] [CrossRef]
- Lengton, R.; Iyer, A.M.; van der Valk, E.S.; Hoogeveen, E.K.; Meijer, O.C.; van der Voorn, B.; van Rossum, E. Variation in glucocorticoid sensitivity and the relation with obesity. Obes. Rev. 2022, 23, e13401. [Google Scholar] [CrossRef] [PubMed]
- Budzyńska, A.; Marek, T.; Nowak, A.; Kaczor, R.; Nowakowska-Dulawa, E. A prospective, randomized, placebo-controlled trial of prednisone and allopurinol in the prevention of ERCP-induced pancreatitis. Endoscopy 2001, 33, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Cooke, W.T.; Meynell, M.J.; Rogers, N.C.; Wilson, A.O. Treatment of acute pancreatitis with cortisone. Lancet 1956, 271, 651–652. [Google Scholar] [PubMed]
- Cosen-Binker, L.I.; Binker, M.G.; Cosen, R.; Negri, G.; Tiscornia, O. Influence of hydrocortisone, prednisolone, and NO association on the evolution of acute pancreatitis. Dig. Dis. Sci. 2006, 51, 915–925. [Google Scholar] [CrossRef]
- Dumot, J.A.; Conwell, D.L.; O’Connor, J.B.; Ferguson, D.R.; Vargo, J.J.; Barnes, D.S.; Shay, S.S.; Sterling, M.J.; Horth, K.S.; Issa, K.; et al. Pretreatment with methylprednisolone to prevent ERCP-induced pancreatitis: A randomized, multicenter, placebo-controlled clinical trial. Am. J. Gastroenterol. 1998, 93, 61–65. [Google Scholar] [CrossRef]
- Wang, Z.F.; Liu, C.; Lu, Y.; Dong, R.; Xu, J.; Yu, L.; Yao, Y.M.; Liu, Q.G.; Pan, C.E. Dexamethasone and dextran 40 treatment of 32 patients with severe acute pancreatitis. World J. Gastroenterol. 2004, 10, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.E.; Coutinho, A.E.; Zhang, Z.; Kipari, T.; Savill, J.S.; Seckl, J.R. Changing glucocorticoid action: 11β-hydroxysteroid dehydrogenase type 1 in acute and chronic inflammation. J. Steroid Biochem. Mol. Biol. 2013, 137, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley-Yates, P.T. Inhaled corticosteroids: Potency, dose equivalence and therapeutic index. Br. J. Clin. Pharmacol. 2015, 80, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, S.; Eigendorff, E.; Burkhardt, P.; Quinkler, M.; Bumke-Vogt, C.; Rochel, M.; Seidelmann, D.; Esperling, P.; Oelkers, W.; Bähr, V. 11beta-hydroxysteroid dehydrogenase types 1 and 2: An important pharmacokinetic determinant for the activity of synthetic mineralo- and glucocorticoids. J. Clin. Endocrinol. Metab. 2002, 87, 5695–5701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharmacokinet. 2005, 44, 61–98. [Google Scholar] [CrossRef]
- Hall, E.D. The neuroprotective pharmacology of methylprednisolone. J. Neurosurg. 1992, 76, 13–22. [Google Scholar] [CrossRef]
- Rodríguez Villanueva, J.; Rodríguez Villanueva, L.; Guzmán Navarro, M. Pharmaceutical technology can turn a traditional drug, dexamethasone into a first-line ocular medicine. A global perspective and future trends. Int. J. Pharm. 2017, 516, 342–351. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Salt, A.N.; Plontke, S.K. Pharmacokinetic principles in the inner ear: Influence of drug properties on intratympanic applications. Hear. Res. 2018, 368, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Zen, M.; Canova, M.; Campana, C.; Bettio, S.; Nalotto, L.; Rampudda, M.; Ramonda, R.; Iaccarino, L.; Doria, A. The kaleidoscope of glucorticoid effects on immune system. Autoimmun. Rev. 2011, 10, 305–310. [Google Scholar] [CrossRef]
- Zhang, M.; Moore, G.A.; Jensen, B.P.; Begg, E.J.; Bird, P.A. Determination of dexamethasone and dexamethasone sodium phosphate in human plasma and cochlear perilymph by liquid chromatography/tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 17–24. [Google Scholar] [CrossRef]
- Petrov, M.S.; Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 175–184. [Google Scholar] [CrossRef]
- Fagenholz, P.J.; Fernández-del Castillo, C.; Harris, N.S.; Pelletier, A.J.; Camargo, C.A., Jr. Direct medical costs of acute pancreatitis hospitalizations in the United States. Pancreas 2007, 35, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Luo, Y.; Okoye, C.S.; Chen, H.; Liu, J.; Zhang, G.; Xu, C.; Chen, H. Intestinal barrier damage, systemic inflammatory response syndrome, and acute lung injury: A troublesome trio for acute pancreatitis. Biomed. Pharmacother 2020, 132, 110770. [Google Scholar] [CrossRef] [PubMed]
- Hietaranta, A.; Kemppainen, E.; Puolakkainen, P.; Sainio, V.; Haapiainen, R.; Peuravuori, H.; Kivilaakso, E.; Nevalainen, T. Extracellular phospholipases A2 in relation to systemic inflammatory response syndrome (SIRS) and systemic complications in severe acute pancreatitis. Pancreas 1999, 18, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Frausto, M.S.; Pittet, D.; Costigan, M.; Hwang, T.; Davis, C.S.; Wenzel, R.P. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA 1995, 273, 117–123. [Google Scholar] [CrossRef]
- Bhatia, M. Novel therapeutic targets for acute pancreatitis and associated multiple organ dysfunction syndrome. Curr. Drug Targets-Inflamm. Allergy 2002, 1, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. Inflammatory response on the pancreatic acinar cell injury. Scand. J. Surg. 2005, 94, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, M.; Brady, M.; Shokuhi, S.; Christmas, S.; Neoptolemos, J.P.; Slavin, J. Inflammatory mediators in acute pancreatitis. J. Pathol. 2000, 190, 117–125. [Google Scholar] [CrossRef]
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 2004, 202, 145–156. [Google Scholar] [CrossRef]
- Bhatia, M.; Wong, F.L.; Cao, Y.; Lau, H.Y.; Huang, J.; Puneet, P.; Chevali, L. Pathophysiology of acute pancreatitis. Pancreatology 2005, 5, 132–144. [Google Scholar] [CrossRef]
- Adib-Conquy, M.; Cavaillon, J.M. Compensatory anti-inflammatory response syndrome. Thromb. Haemost. 2009, 101, 36–47. [Google Scholar]
- Xiong, J.; Zhu, S.; Zhou, Y.; Wu, H.; Wang, C. Regulation of omega-3 fish oil emulsion on the SIRS during the initial stage of severe acute pancreatitis. J. Huazhong Univ. Sci. Technol. 2009, 29, 35–38. [Google Scholar] [CrossRef]
- Ward, N.S.; Casserly, B.; Ayala, A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin. Chest Med. 2008, 29, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bone, R.C. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit. Care Med. 1996, 24, 1125–1128. [Google Scholar] [CrossRef]
- Gunjaca, I.; Zunic, J.; Gunjaca, M.; Kovac, Z. Circulating cytokine levels in acute pancreatitis-model of SIRS/CARS can help in the clinical assessment of disease severity. Inflammation 2012, 35, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Osuchowski, M.F.; Welch, K.; Siddiqui, J.; Remick, D.G. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J. Immunol. 2006, 177, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Zhang, L.; Chen, L.J.; Cheng, Q.H.; Wang, J.M.; Cai, W.; Shen, H.P.; Cai, J. Influence of dexamethasone on inflammatory mediators and NF-kappaB expression in multiple organs of rats with severe acute pancreatitis. World J. Gastroenterol. 2007, 13, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.P.; Zhang, L.; Wang, Y.; Cheng, Q.H.; Wang, J.M.; Cai, W.; Shen, H.P.; Cai, J. Study of the protective effects of dexamethasone on multiple organ injury in rats with severe acute pancreatitis. Jop 2007, 8, 400–412. [Google Scholar]
- Zhao, Y.; Xiong, R.P.; Chen, X.; Li, P.; Ning, Y.L.; Yang, N.; Peng, Y.; Jiang, Y.L.; Zhou, Y.G. Hsp90 regulation affects the treatment of glucocorticoid for pancreatitis-induced lung injury. Mol. Cell. Biochem. 2018, 440, 189–197. [Google Scholar] [CrossRef]
- Kimura, K.; Shimosegawa, T.; Sasano, H.; Abe, R.; Satoh, A.; Masamune, A.; Koizumi, M.; Nagura, H.; Toyota, T. Endogenous glucocorticoids decrease the acinar cell sensitivity to apoptosis during cerulein pancreatitis in rats. Gastroenterology 1998, 114, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Shimosegawa, T.; Kimura, K.; Abe, T.; Kashimura, J.; Koizumi, M.; Toyota, T. The role of endogenous glucocorticoids in rat experimental models of acute pancreatitis. Gastroenterology 1995, 109, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.A.; Vogeser, M.; Belyaev, O.; Gloor, B.; Strobel, O.; Weyhe, D.; Werner, J.; Borgstrom, A.; Buchler, M.W.; Uhl, W. Role of endogenous glucocorticoid metabolism in human acute pancreatitis. Crit. Care Med. 2006, 34, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, G.K.; Jurutka, P.W.; Haussler, C.A.; Haussler, M.R. Steroid hormone receptors: Evolution, ligands, and molecular basis of biologic function. J. Cell. Biochem. 1999, 75 (Suppl. 32–33), 110–122. [Google Scholar] [CrossRef]
- Frank, F.; Ortlund, E.A.; Liu, X. Structural insights into glucocorticoid receptor function. Biochem. Soc. Trans. 2021, 49, 2333–2343. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Busillo, J.M.; Cidlowski, J.A. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab. 2013, 24, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Beck, I.M.; De Bosscher, K.; Haegeman, G. Glucocorticoid receptor mutants: Man-made tools for functional research. Trends Endocrinol. Metab. 2011, 22, 295–310. [Google Scholar] [CrossRef]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [PubMed] [Green Version]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br. J. Pharmacol. 2006, 148, 245–254. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Nissen, R.M.; Yamamoto, K.R. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000, 14, 2314–2329. [Google Scholar] [CrossRef] [Green Version]
- Yang-Yen, H.F.; Chambard, J.C.; Sun, Y.L.; Smeal, T.; Schmidt, T.J.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215. [Google Scholar] [CrossRef]
- Eychène, A.; Rocques, N.; Pouponnot, C. A new MAFia in cancer. Nat. Rev. Cancer 2008, 8, 683–693. [Google Scholar] [CrossRef]
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2008, 10, 201. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Schüle, R.; Rangarajan, P.; Kliewer, S.; Ransone, L.J.; Bolado, J.; Yang, N.; Verma, I.M.; Evans, R.M. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell 1990, 62, 1217–1226. [Google Scholar] [CrossRef]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- Koj, A. Initiation of acute phase response and synthesis of cytokines. Biochim. Biophys. Acta 1996, 1317, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Roebuck, K.A.; Carpenter, L.R.; Lakshminarayanan, V.; Page, S.M.; Moy, J.N.; Thomas, L.L. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J. Leukoc. Biol. 1999, 65, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Zagariya, A.; Mungre, S.; Lovis, R.; Birrer, M.; Ness, S.; Thimmapaya, B.; Pope, R. Tumor necrosis factor alpha gene regulation: Enhancement of C/EBPbeta-induced activation by c-Jun. Mol. Cell. Biol. 1998, 18, 2815–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.F.; Lentsch, A.B.; Sarma, J.V.; Sun, L.; Riedemann, N.C.; McClintock, S.D.; McGuire, S.R.; Van Rooijen, N.; Ward, P.A. Activator protein-1 activation in acute lung injury. Am. J. Pathol. 2002, 161, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentsch, A.B.; Czermak, B.J.; Bless, N.M.; Ward, P.A. NF-kappaB activation during IgG immune complex-induced lung injury: Requirements for TNF-alpha and IL-1beta but not complement. Am. J. Pathol. 1998, 152, 1327–1336. [Google Scholar] [PubMed]
- Cao, L.; Qian, L.L.; Zhu, Y.R.; Guo, C.B.; Gong, X.H.; Sun, B. Regulation of activity of nuclear factor-kappaB and activator protein-1 by nitric oxide, surfactant and glucocorticoids in alveolar macrophages from piglets with acute lung injury. Acta Pharmacol. Sin. 2003, 24, 1316–1323. [Google Scholar]
- Chinenov, Y.; Rogatsky, I. Glucocorticoids and the innate immune system: Crosstalk with the toll-like receptor signaling network. Mol. Cell. Endocrinol. 2007, 275, 30–42. [Google Scholar] [CrossRef]
- Kassel, O.; Herrlich, P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol. Cell. Endocrinol. 2007, 275, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, A.C.; Druker, J.; Refojo, D.; Holsboer, F.; Arzt, E. Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FASEB J. 2009, 23, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Liberman, A.C.; Refojo, D.; Druker, J.; Toscano, M.; Rein, T.; Holsboer, F.; Arzt, E. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J. 2007, 21, 1177–1188. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Lozach, J.; Benner, C.; Pascual, G.; Tangirala, R.K.; Westin, S.; Hoffmann, A.; Subramaniam, S.; David, M.; Rosenfeld, M.G.; et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell 2005, 122, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Reily, M.M.; Pantoja, C.; Hu, X.; Chinenov, Y.; Rogatsky, I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. 2006, 25, 108–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.D.; Zhu, R.X.; Bian, Z.Z.; Pan, X.T. Improvement of Gut Microbiota by Inhibition of P38 Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway in Rats with Severe Acute Pancreatitis. Med. Sci. Monit. 2019, 25, 4609–4616. [Google Scholar] [CrossRef]
- Papachristou, D.J.; Papadakou, E.; Basdra, E.K.; Baltopoulos, P.; Panagiotopoulos, E.; Papavassiliou, A.G. Involvement of the p38 MAPK-NF-kappaB signal transduction pathway and COX-2 in the pathobiology of meniscus degeneration in humans. Mol. Med. 2008, 14, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.; Sully, G.; Clark, A.R.; Saklatvala, J. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett. 2000, 483, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.L.; Brook, M.; Clark, A.R.; Saklatvala, J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J. Biol. Chem. 1999, 274, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Lasa, M.; Mahtani, K.R.; Finch, A.; Brewer, G.; Saklatvala, J.; Clark, A.R. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol. Cell. Biol. 2000, 20, 4265–4274. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, K.; Mori, A.; Miyata, H.; Akahane, M.; Ajisawa, Y.; Okudaira, H. Regulation of interleukin-1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 24832–24838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagès, G.; Berra, E.; Milanini, J.; Levy, A.P.; Pouysségur, J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J. Biol. Chem. 2000, 275, 26484–26491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, S.H.; Dean, J.L.; Sarsfield, S.J.; Brook, M.; Clark, A.R.; Saklatvala, J. A p38 MAP kinase inhibitor regulates stability of interleukin-1-induced cyclooxygenase-2 mRNA. FEBS Lett. 1998, 439, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Treisman, R. Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol. 1996, 8, 205–215. [Google Scholar] [CrossRef]
- Wang, S.W.; Pawlowski, J.; Wathen, S.T.; Kinney, S.D.; Lichenstein, H.S.; Manthey, C.L. Cytokine mRNA decay is accelerated by an inhibitor of p38-mitogen-activated protein kinase. Inflamm. Res. 1999, 48, 533–538. [Google Scholar] [CrossRef]
- Winzen, R.; Kracht, M.; Ritter, B.; Wilhelm, A.; Chen, C.Y.; Shyu, A.B.; Müller, M.; Gaestel, M.; Resch, K.; Holtmann, H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999, 18, 4969–4980. [Google Scholar] [CrossRef]
- Nick, J.A.; Young, S.K.; Arndt, P.G.; Lieber, J.G.; Suratt, B.T.; Poch, K.R.; Avdi, N.J.; Malcolm, K.C.; Taube, C.; Henson, P.M.; et al. Selective suppression of neutrophil accumulation in ongoing pulmonary inflammation by systemic inhibition of p38 mitogen-activated protein kinase. J. Immunol. 2002, 169, 5260–5269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; Smythe, C.; Keyse, S.M. The human CL100 gene encodes a Tyr/Thr-protein phosphatase which potently and specifically inactivates MAP kinase and suppresses its activation by oncogenic ras in Xenopus oocyte extracts. Oncogene 1993, 8, 2015–2020. [Google Scholar] [PubMed]
- Hutter, D.; Chen, P.; Barnes, J.; Liu, Y. Catalytic activation of mitogen-activated protein (MAP) kinase phosphatase-1 by binding to p38 MAP kinase: Critical role of the p38 C-terminal domain in its negative regulation. Biochem. J. 2000, 352 Pt 1, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; New, L.; Han, J.; Molkentin, J.D. Calcineurin enhances MAPK phosphatase-1 expression and p38 MAPK inactivation in cardiac myocytes. J. Biol. Chem. 2001, 276, 15913–15919. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Charles, C.H.; Lau, L.F.; Tonks, N.K. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993, 75, 487–493. [Google Scholar] [CrossRef]
- Lasa, M.; Abraham, S.M.; Boucheron, C.; Saklatvala, J.; Clark, A.R. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell. Biol. 2002, 22, 7802–7811. [Google Scholar]
- Antonicelli, F.; De Coupade, C.; Russo-Marie, F.; Le Garrec, Y. CREB is involved in mouse annexin A1 regulation by cAMP and glucocorticoids. Eur. J. Biochem. 2001, 268, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Rhee, H.J.; Ko, J.; Kim, Y.J.; Kim, H.G.; Yang, J.M.; Choi, E.C.; Na, D.S. Inhibition of cytosolic phospholipase A2 by annexin I. Specific interaction model and mapping of the interaction site. J. Biol. Chem. 2001, 276, 15712–15719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, H.; Uemura, K.; Moriyama, A.; Wada, Y.; Asai, K.; Kimura, S.; Kato, T. Glucocorticoid induced the expression of mRNA and the secretion of lipocortin 1 in rat astrocytoma cells. Brain Res. 1997, 746, 256–264. [Google Scholar] [CrossRef]
- Solito, E.; de Coupade, C.; Parente, L.; Flower, R.J.; Russo-Marie, F. IL-6 stimulates annexin 1 expression and translocation and suggests a new biological role as class II acute phase protein. Cytokine 1998, 10, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Isaji, S.; Hayashi, J.; Higashiguchi, T.; Yokoi, H.; Ogura, Y.; Noguchi, T.; Kawarada, Y. Effect of IS-741 (a new synthetic antiinflammatory agent) on acute necrotizing pancreatitis in dogs. Significance of its inhibitory effect on cytosolic phospholipase A2. Digestion 1999, 60 (Suppl. 1), 47–51. [Google Scholar] [CrossRef]
- Amandi-Burgermeister, E.; Tibes, U.; Kaiser, B.M.; Friebe, W.G.; Scheuer, W.V. Suppression of cytokine synthesis, integrin expression and chronic inflammation by inhibitors of cytosolic phospholipase A2. Eur. J. Pharmacol. 1997, 326, 237–250. [Google Scholar] [CrossRef]
- Hayashi, J.; Kawarada, Y.; Isaji, S.; Yokoi, H.; Higashiguchi, T. Therapeutic effects of continuous intraarterial antibiotic infusion in preventing pancreatic infection in experimental acute necrotizing pancreatitis. Pancreas 1996, 13, 184–192. [Google Scholar] [CrossRef]
- Nagase, T.; Uozumi, N.; Ishii, S.; Kume, K.; Izumi, T.; Ouchi, Y.; Shimizu, T. Acute lung injury by sepsis and acid aspiration: A key role for cytosolic phospholipase A2. Nat. Immunol. 2000, 1, 42–46. [Google Scholar] [CrossRef]
- Lim, L.H.; Solito, E.; Russo-Marie, F.; Flower, R.J.; Perretti, M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: Effect of lipocortin 1. Proc. Natl. Acad. Sci. USA 1998, 95, 14535–14539. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Pipitone, N.; Perretti, M. Regulation of leukocyte-endothelial interactions by glucocorticoids. Ann. N. Y. Acad. Sci. 2002, 966, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Roviezzo, F.; Getting, S.J.; Paul-Clark, M.J.; Yona, S.; Gavins, F.N.; Perretti, M.; Hannon, R.; Croxtall, J.D.; Buckingham, J.C.; Flower, R.J. The annexin-1 knockout mouse: What it tells us about the inflammatory response. J. Physiol. Pharmacol. 2002, 53, 541–553. [Google Scholar] [PubMed]
- Ethridge, R.T.; Chung, D.H.; Slogoff, M.; Ehlers, R.A.; Hellmich, M.R.; Rajaraman, S.; Saito, H.; Uchida, T.; Evers, B.M. Cyclooxygenase-2 gene disruption attenuates the severity of acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology 2002, 123, 1311–1322. [Google Scholar] [CrossRef]
- Alaaeddine, N.; Di Battista, J.A.; Pelletier, J.P.; Kiansa, K.; Cloutier, J.M.; Martel-Pelletier, J. Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: Distinct targeting in the signaling pathways. Arthritis Rheum. 1999, 42, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Kuitert, L.M.; Bergmann, M.; Adcock, I.M.; Barnes, P.J. Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem. Biophys. Res. Commun. 1997, 237, 28–32. [Google Scholar] [CrossRef]
- Niiro, H.; Otsuka, T.; Ogami, E.; Yamaoka, K.; Nagano, S.; Akahoshi, M.; Nakashima, H.; Arinobu, Y.; Izuhara, K.; Niho, Y. MAP kinase pathways as a route for regulatory mechanisms of IL-10 and IL-4 which inhibit COX-2 expression in human monocytes. Biochem. Biophys. Res. Commun. 1998, 250, 200–205. [Google Scholar] [CrossRef]
- Reddy, S.T.; Gilbert, R.S.; Xie, W.; Luner, S.; Herschman, H.R. TGF-beta 1 inhibits both endotoxin-induced prostaglandin synthesis and expression of the TIS10/prostaglandin synthase 2 gene in murine macrophages. J. Leukoc. Biol. 1994, 55, 192–200. [Google Scholar] [CrossRef]
- Inoue, H.; Umesono, K.; Nishimori, T.; Hirata, Y.; Tanabe, T. Glucocorticoid-mediated suppression of the promoter activity of the cyclooxygenase-2 gene is modulated by expression of its receptor in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1999, 254, 292–298. [Google Scholar] [CrossRef]
- Tanabe, T.; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar] [CrossRef]
- Cannarile, L.; Zollo, O.; D’Adamio, F.; Ayroldi, E.; Marchetti, C.; Tabilio, A.; Bruscoli, S.; Riccardi, C. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001, 8, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Morand, E.; Yang, Y.H. Development of novel treatment strategies for inflammatory diseases-similarities and divergence between glucocorticoids and GILZ. Front. Pharmacol. 2014, 5, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingawale, D.K.; Mandlik, S.K. New insights into the novel anti-inflammatory mode of action of glucocorticoids. Immunopharmacol. Immunotoxicol. 2020, 42, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R.T.; Hoppstädter, J.; Hirschfelder, K.; Hachenthal, N.; Diesel, B.; Kessler, S.M.; Huwer, H.; Kiemer, A.K. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis 2014, 234, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Carrick, D.M.; Lai, W.S.; Blackshear, P.J. The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis Res. Ther. 2004, 6, 248–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrestensen, C.A.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F.; Pelo, J.W.; Worthington, M.T.; Sturgill, T.W. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J. Biol. Chem. 2004, 279, 10176–10184. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.F.; Sapolsky, R.M. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav. Immun. 2007, 21, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, S.E.; Firestein, G.S. Mitogen activated protein kinase inhibitors: Where are we now and where are we going. Ann. Rheum. Dis. 2006, 65 (Suppl. 3), iii83–iii88. [Google Scholar] [CrossRef]
- Beck, I.M.; Vanden Berghe, W.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in inflammation: The interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [Green Version]
- Beck, I.M.; Vanden Berghe, W.; Vermeulen, L.; Bougarne, N.; Vander Cruyssen, B.; Haegeman, G.; De Bosscher, K. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J. 2008, 27, 1682–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, Y.; Tomita, K.; Watanabe, M.; Yamasaki, A.; Sano, H.; Hitsuda, Y.; Shimizu, E. Dexamethasone inhibits phosphorylation of histone H3 at serine 10. Biochem. Biophys. Res. Commun. 2005, 336, 1049–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, G.C.; Deterding, L.J.; Tomer, K.B.; Archer, T.K. Hormone-mediated dephosphorylation of specific histone H1 isoforms. J. Biol. Chem. 2001, 276, 36467–36473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, R.N.; Banks, G.C.; Trotter, K.W.; Lee, H.L.; Archer, T.K. Histone H1 phosphorylation by Cdk2 selectively modulates mouse mammary tumor virus transcription through chromatin remodeling. Mol. Cell. Biol. 2001, 21, 5417–5425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, H.H.; Cram, E.J.; Wang, E.C.; Huang, A.J.; Kasler, H.G.; Firestone, G.L. Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J. Biol. Chem. 1998, 273, 1998–2007. [Google Scholar] [CrossRef] [Green Version]
- Cram, E.J.; Ramos, R.A.; Wang, E.C.; Cha, H.H.; Nishio, Y.; Firestone, G.L. Role of the CCAAT/enhancer binding protein-alpha transcription factor in the glucocorticoid stimulation of p21waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J. Biol. Chem. 1998, 273, 2008–2014. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, A.K.; Hu, J.; Basu, S.; Hay, J.; Reibman, J.; Yie, T.A.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am. J. Respir. Cell Mol. Biol. 2002, 27, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Rogatsky, I.; Trowbridge, J.M.; Garabedian, M.J. Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol. Cell. Biol. 1997, 17, 3181–3193. [Google Scholar] [CrossRef] [Green Version]
- Gloire, G.; Horion, J.; El Mjiyad, N.; Bex, F.; Chariot, A.; Dejardin, E.; Piette, J. Promoter-dependent effect of IKKalpha on NF-kappaB/p65 DNA binding. J. Biol. Chem. 2007, 282, 21308–21318. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.R. Anti-inflammatory functions of glucocorticoid-induced genes. Mol. Cell. Endocrinol. 2007, 275, 79–97. [Google Scholar] [CrossRef] [Green Version]
- De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: Molecular mechanisms for gene repression. Endocr. Rev. 2003, 24, 488–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- McCoy, C.E.; Carpenter, S.; Pålsson-McDermott, E.M.; Gearing, L.J.; O’Neill, L.A. Glucocorticoids inhibit IRF3 phosphorylation in response to Toll-like receptor-3 and -4 by targeting TBK1 activation. J. Biol. Chem. 2008, 283, 14277–14285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Z.; Yu, Z.; Xia, G.; Wang, Y.; Wang, L.; Peng, C.; Jiang, B.; Liu, S. Wip1 Aggravates the Cerulein-Induced Cell Autophagy and Inflammatory Injury by Targeting STING/TBK1/IRF3 in Acute Pancreatitis. Inflammation 2021, 44, 1175–1183. [Google Scholar] [CrossRef]
- Abu-Amer, Y.; Ross, F.P.; McHugh, K.P.; Livolsi, A.; Peyron, J.F.; Teitelbaum, S.L. Tumor necrosis factor-alpha activation of nuclear transcription factor-kappaB in marrow macrophages is mediated by c-Src tyrosine phosphorylation of Ikappa Balpha. J. Biol. Chem. 1998, 273, 29417–29423. [Google Scholar] [CrossRef] [Green Version]
- Mong, P.Y.; Wang, Q. Activation of Rho kinase isoforms in lung endothelial cells during inflammation. J. Immunol. 2009, 182, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, N.M.; Callahan, J.A.; Lo, D.H.; Firestone, G.L. Selective glucocorticoid control of Rho kinase isoforms regulate cell-cell interactions. Biochem. Biophys. Res. Commun. 2007, 354, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Cato, A.C.; Nestl, A.; Mink, S. Rapid actions of steroid receptors in cellular signaling pathways. Sci. STKE Signal Transduct. Knowl. Environ. 2002, 2002, re9. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Choudhury, Q.; Flower, R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000, 130, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joëls, M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol. Cell. Endocrinol. 2012, 350, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, R.A.; Witchell, S.F.; DeFranco, D.B. Cooperativity and complementarity: Synergies in non-classical and classical glucocorticoid signaling. Cell Cycle 2012, 11, 2819–2827. [Google Scholar] [CrossRef] [Green Version]
- Stahn, C.; Löwenberg, M.; Hommes, D.W.; Buttgereit, F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol. Cell. Endocrinol. 2007, 275, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Buttgereit, F.; Scheffold, A. Rapid glucocorticoid effects on immune cells. Steroids 2002, 67, 529–534. [Google Scholar] [CrossRef]
- Buttgereit, F.; Straub, R.H.; Wehling, M.; Burmester, G.R. Glucocorticoids in the treatment of rheumatic diseases: An update on the mechanisms of action. Arthritis Rheum. 2004, 50, 3408–3417. [Google Scholar] [CrossRef]
- Bartholome, B.; Spies, C.M.; Gaber, T.; Schuchmann, S.; Berki, T.; Kunkel, D.; Bienert, M.; Radbruch, A.; Burmester, G.R.; Lauster, R.; et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. 2004, 18, 70–80. [Google Scholar] [CrossRef]
- Gametchu, B.; Chen, F.; Sackey, F.; Powell, C.; Watson, C.S. Plasma membrane-resident glucocorticoid receptors in rodent lymphoma and human leukemia models. Steroids 1999, 64, 107–119. [Google Scholar] [CrossRef]
- Tomkötter, L.; Erbes, J.; Trepte, C.; Hinsch, A.; Dupree, A.; Bockhorn, M.; Mann, O.; Izbicki, J.R.; Bachmann, K. The Effects of Pancreatic Microcirculatory Disturbances on Histopathologic Tissue Damage and the Outcome in Severe Acute Pancreatitis. Pancreas 2016, 45, 248–253. [Google Scholar] [CrossRef]
- Cuthbertson, C.M.; Christophi, C. Disturbances of the microcirculation in acute pancreatitis. Br. J. Surg. 2006, 93, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, M.; Yamauchi, J.; Shibuya, K.; Chen, H.M.; Ding, L.; Takeda, K.; Kobari, M.; Matsuno, S. Pancreatic microcirculation in acute pancreatitis. J. Hepato-Biliary-Pancreat. Surg. 1998, 5, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kinnala, P.J.; Kuttila, K.T.; Grönroos, J.M.; Havia, T.V.; Nevalainen, T.J.; Niinikoski, J.H. Pancreatic tissue perfusion in experimental acute pancreatitis. Eur. J. Surg. 2001, 167, 689–694. [Google Scholar] [PubMed]
- Spormann, H.; Sokolowski, A.; Letko, G. Effect of temporary ischemia upon development and histological patterns of acute pancreatitis in the rat. Pathol. Res. Pract. 1989, 184, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, B.; Menger, M.D. Microcirculatory dysfunction in acute pancreatitis. A new concept of pathogenesis involving vasomotion-associated arteriolar constriction and dilation. Pancreatology 2003, 3, 181–190. [Google Scholar] [CrossRef]
- Granell, S.; Bulbena, O.; Genesca, M.; Sabater, L.; Sastre, J.; Gelpi, E.; Closa, D. Mobilization of xanthine oxidase from the gastrointestinal tract in acute pancreatitis. BMC Gastroenterol. 2004, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Schoenberg, M.H.; Büchler, M.; Gaspar, M.; Stinner, A.; Younes, M.; Melzner, I.; Bültmann, B.; Beger, H.G. Oxygen free radicals in acute pancreatitis of the rat. Gut 1990, 31, 1138–1143. [Google Scholar] [CrossRef] [Green Version]
- Frossard, J.L.; Saluja, A.; Bhagat, L.; Lee, H.S.; Bhatia, M.; Hofbauer, B.; Steer, M.L. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology 1999, 116, 694–701. [Google Scholar] [CrossRef]
- Poch, B.; Gansauge, F.; Rau, B.; Wittel, U.; Gansauge, S.; Nüssler, A.K.; Schoenberg, M.; Beger, H.G. The role of polymorphonuclear leukocytes and oxygen-derived free radicals in experimental acute pancreatitis: Mediators of local destruction and activators of inflammation. FEBS Lett. 1999, 461, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Rau, B.; Bauer, A.; Wang, A.; Gansauge, F.; Weidenbach, H.; Nevalainen, T.; Poch, B.; Beger, H.G.; Nussler, A.K. Modulation of endogenous nitric oxide synthase in experimental acute pancreatitis: Role of anti-ICAM-1 and oxygen free radical scavengers. Ann. Surg. 2001, 233, 195–203. [Google Scholar] [CrossRef]
- Werner, J.; Z’graggen, K.; Fernández-del Castillo, C.; Lewandrowski, K.B.; Compton, C.C.; Warshaw, A.L. Specific therapy for local and systemic complications of acute pancreatitis with monoclonal antibodies against ICAM-1. Ann. Surg. 1999, 229, 834–840; discussion 841–842. [Google Scholar] [CrossRef]
- Mimidis, K.; Papadopoulos, V.; Kartasis, Z.; Baka, M.; Tsatlidis, V.; Bourikas, G.; Kartalis, G. Assessment of platelet adhesiveness and aggregation in mild acute pancreatitis using the PFA-100 system. Jop 2004, 5, 132–137. [Google Scholar] [PubMed]
- Ranson, J.H.; Lackner, H.; Berman, I.R.; Schinella, R. The relationship of coagulation factors to clinical complications of acute pancreatitis. Surgery 1977, 81, 502–511. [Google Scholar] [PubMed]
- Salomone, T.; Tosi, P.; Palareti, G.; Tomassetti, P.; Migliori, M.; Guariento, A.; Saieva, C.; Raiti, C.; Romboli, M.; Gullo, L. Coagulative disorders in human acute pancreatitis: Role for the D-dimer. Pancreas 2003, 26, 111–116. [Google Scholar] [CrossRef]
- Kelly, D.M.; McEntee, G.P.; McGeeney, K.F.; Fitzpatrick, J.M. Microvasculature of the pancreas, liver, and kidney in cerulein-induced pancreatitis. Arch. Surg. 1993, 128, 293–295. [Google Scholar] [CrossRef]
- McEntee, G.; Leahy, A.; Cottell, D.; Dervan, P.; McGeeney, K.; Fitzpatrick, J.M. Three-dimensional morphological study of the pancreatic microvasculature in caerulein-induced experimental pancreatitis. Br. J. Surg. 1989, 76, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Schiller, W.R.; Anderson, M.C. Microcirculation of the normal and inflamed canine pancreas. Ann. Surg. 1975, 181, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Weidenbach, H.; Lerch, M.M.; Gress, T.M.; Pfaff, D.; Turi, S.; Adler, G. Vasoactive mediators and the progression from oedematous to necrotising experimental acute pancreatitis. Gut 1995, 37, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.M.; McEntee, G.P.; Delaney, C.; McGeeney, K.F.; Fitzpatrick, J.M. Temporal relationship of acinar and microvascular changes in caerulein-induced pancreatitis. Br. J. Surg. 1993, 80, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Pitkäranta, P.; Kivisaari, L.; Nordling, S.; Nuutinen, P.; Schroder, T. Vascular changes of pancreatic ducts and vessels in acute necrotizing, and in chronic pancreatitis in humans. Int. J. Pancreatol. 1991, 8, 13–22. [Google Scholar] [CrossRef]
- Wright, P.W.; Goodhead, B. Prevention of hemorrhagic pancreatitis with fibrinolysin or heparin. Arch. Surg. 1970, 100, 42–46. [Google Scholar] [CrossRef]
- Anderson, M.C.; Schoenfeld, F.B.; Iams, W.B.; Suwa, M. Circulatory changes in acute pancreatitis. Surg. Clin. N. Am. 1967, 47, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Eibl, G.; Buhr, H.J.; Foitzik, T. Therapy of microcirculatory disorders in severe acute pancreatitis: What mediators should we block. Intensive Care Med. 2002, 28, 139–146. [Google Scholar] [CrossRef]
- Kalter, E.S.; Carlson, R.W.; Thijs, L.G.; Weil, M.H. Effects of methylprednisolone on hemodynamics, arteriovenous oxygen difference, P50, and 2,3 DPG in bacterial shock: A preliminary study. Crit. Care Med. 1982, 10, 662–666. [Google Scholar] [CrossRef]
- Lillehei, R.C.; Longerbeam, J.K.; Bloch, J.H.; Manax, W.G. The modern treatment of shock based on physiologic principles. Clin. Pharmacol. Ther. 1964, 5, 63–101. [Google Scholar] [CrossRef]
- Studley, J.G.; Schenk, W.G., Jr. Pathophysiology of acute pancreatitis: Evaluation of the effect and mode of action of steroids in experimental pancreatitis in dogs. Am. J. Surg. 1982, 143, 761–764. [Google Scholar] [CrossRef]
- Laviolle, B.; Nesseler, N.; Massart, C.; Bellissant, E. Fludrocortisone and hydrocortisone, alone or in combination, on in vivo hemodynamics and in vitro vascular reactivity in normal and endotoxemic rats: A randomized factorial design study. J. Cardiovasc. Pharmacol. 2014, 63, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Kimmel, S.C.; Levin, R.I.; Martiniuk, F.; Weissmann, G. A mechanism for the antiinflammatory effects of corticosteroids: The glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1992, 89, 9991–9995. [Google Scholar] [CrossRef] [PubMed]
- Davenpeck, K.L.; Zagorski, J.; Schleimer, R.P.; Bochner, B.S. Lipopolysaccharide-induced leukocyte rolling and adhesion in the rat mesenteric microcirculation: Regulation by glucocorticoids and role of cytokines. J. Immunol. 1998, 161, 6861–6870. [Google Scholar] [CrossRef]
- Tailor, A.; Tomlinson, A.; Salas, A.; Panés, J.; Granger, D.N.; Flower, R.J.; Perretti, M. Dexamethasone inhibition of leucocyte adhesion to rat mesenteric postcapillary venules: Role of intercellular adhesion molecule 1 and KC. Gut 1999, 45, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Vachharajani, V.; Vital, S.; Russell, J.; Scott, L.K.; Granger, D.N. Glucocorticoids inhibit the cerebral microvascular dysfunction associated with sepsis in obese mice. Microcirculation 2006, 13, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Thorlacius, H. Dexamethasone inhibits arteriolar leukocyte rolling and adhesion induced by tumor necrosis factor-alpha in vivo. Inflamm. Res. 2000, 49, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Wehling, M.; Burmester, G.R. A new hypothesis of modular glucocorticoid actions: Steroid treatment of rheumatic diseases revisited. Arthritis Rheum. 1998, 41, 761–767. [Google Scholar] [CrossRef]
- Galvin, M.J.; Shupe, K.; Lefer, A.M. Anti-endotoxin actions of methylprednisolone in the isolated perfused cat liver. Pharmacology 1978, 17, 181–190. [Google Scholar] [CrossRef]
- Weissmann, G.; ThomaS, L. Studies on lysosomes. I. The effects of endotoxin, endotoxin tolerance, and cortisone on the release of acid hydrolases from a granular fraction of rabbit liver. J. Exp. Med. 1962, 116, 433–450. [Google Scholar] [CrossRef]
- Zarem, H.A.; Zweifach, B.W. Microcirculatory effects of cortisol. protective action against na4edta damage. Proc. Soc. Exp. Biol. Med. 1965, 118, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Croft, Q.P.; Kalidhar, S.; Brooks, J.T.; Herigstad, M.; Smith, T.G.; Dorrington, K.L.; Robbins, P.A. Dexamethasone mimics aspects of physiological acclimatization to 8 hours of hypoxia but suppresses plasma erythropoietin. J. Appl. Physiol. 2013, 114, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Clerici, C.; Saumon, G. Invited review: Active fluid clearance from the distal air spaces of the lung. J. Appl. Physiol. 2002, 93, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Hori, M.; Sakamoto, K.; Karaki, H.; Ozaki, H. Dexamethasone blocks hypoxia-induced endothelial dysfunction in organ-cultured pulmonary arteries. Am. J. Respir. Crit. Care Med. 2004, 170, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Sartori, C.; Allemann, Y.; Duplain, H.; Lepori, M.; Egli, M.; Lipp, E.; Hutter, D.; Turini, P.; Hugli, O.; Cook, S.; et al. Salmeterol for the prevention of high-altitude pulmonary edema. N. Engl. J. Med. 2002, 346, 1631–1636. [Google Scholar] [CrossRef] [Green Version]
- Meduri, G.U.; Annane, D.; Chrousos, G.P.; Marik, P.E.; Sinclair, S.E. Activation and regulation of systemic inflammation in ARDS: Rationale for prolonged glucocorticoid therapy. Chest 2009, 136, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, A. Alveolitis and collapse in the pathogenesis of pulmonary fibrosis. Am. Rev. Respir. Dis. 1989, 140, 513–524. [Google Scholar] [CrossRef]
- Clark, J.G.; Milberg, J.A.; Steinberg, K.P.; Hudson, L.D. Type III procollagen peptide in the adult respiratory distress syndrome. Association of increased peptide levels in bronchoalveolar lavage fluid with increased risk for death. Ann. Intern. Med. 1995, 122, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.R.; Bernard, G.R.; Brigham, K.L.; Higgins, S.B.; Rinaldo, J.E.; Borovetz, H.S.; Sibbald, W.J.; Kariman, K.; Sprung, C.L. Lung microvascular transport properties measured by multiple indicator dilution methods in patients with adult respiratory distress syndrome. A comparison between patients reversing respiratory failure and those failing to reverse. Am. Rev. Respir. Dis. 1990, 141, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, M.; Yamasawa, F.; Ishizaka, A.; Kato, R.; Kikuchi, K.; Kobayashi, K.; Aoki, T.; Sakamaki, F.; Hasegawa, N.; Kawashiro, T. Serum concentration of 7S collagen and prognosis in patients with the adult respiratory distress syndrome. Thorax 1994, 49, 144–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutor, J.D.; Mason, C.M.; Dobard, E.; Beckerman, R.C.; Summer, W.R.; Nelson, S. Loss of compartmentalization of alveolar tumor necrosis factor after lung injury. Am. J. Respir. Crit. Care Med. 1994, 149, 1107–1111. [Google Scholar] [CrossRef]
- Meduri, G.U. The role of the host defence response in the progression and outcome of ARDS: Pathophysiological correlations and response to glucocorticoid treatment. Eur. Respir. J. 1996, 9, 2650–2670. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, E.J.; DiPietro, L.A. Fibrogenic cytokines and connective tissue production. FASEB J. 1994, 8, 854–861. [Google Scholar] [CrossRef]
- Andrews, C.P.; Coalson, J.J.; Smith, J.D.; Johanson, W.G., Jr. Diagnosis of nosocomial bacterial pneumonia in acute, diffuse lung injury. Chest 1981, 80, 254–258. [Google Scholar] [CrossRef]
- Bell, R.C.; Coalson, J.J.; Smith, J.D.; Johanson, W.G., Jr. Multiple organ system failure and infection in adult respiratory distress syndrome. Ann. Intern. Med. 1983, 99, 293–298. [Google Scholar] [CrossRef]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. Am. Coll. Chest Physicians/Soc. Crit. Care Med. Chest 1992, 101, 1644–1655. [Google Scholar]
- Mancebo, J.; Artigas, A. A clinical study of the adult respiratory distress syndrome. Crit. Care Med. 1987, 15, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U. Late adult respiratory distress syndrome. New Horiz. 1993, 1, 563–577. [Google Scholar]
- Montgomery, A.B.; Stager, M.A.; Carrico, C.J.; Hudson, L.D. Causes of mortality in patients with the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1985, 132, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Seidenfeld, J.J.; Pohl, D.F.; Bell, R.C.; Harris, G.D.; Johanson, W.G., Jr. Incidence, site, and outcome of infections in patients with the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1986, 134, 12–16. [Google Scholar]
- Suchyta, M.R.; Clemmer, T.P.; Elliott, C.G.; Orme, J.F., Jr.; Weaver, L.K. The adult respiratory distress syndrome. A report of survival and modifying factors. Chest 1992, 101, 1074–1079. [Google Scholar] [CrossRef]
- Kollef, M.H.; Schuster, D.P. The acute respiratory distress syndrome. N. Engl. J. Med. 1995, 332, 27–37. [Google Scholar] [CrossRef]
- Ashbaugh, D.G.; Maier, R.V. Idiopathic pulmonary fibrosis in adult respiratory distress syndrome. Diagnosis and treatment. Arch. Surg. 1985, 120, 530–535. [Google Scholar] [CrossRef]
- Hooper, R.G.; Kearl, R.A. Established ARDS treated with a sustained course of adrenocortical steroids. Chest 1990, 97, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meduri, G.U.; Belenchia, J.M.; Estes, R.J.; Wunderink, R.G.; el Torky, M.; Leeper, K.V., Jr. Fibroproliferative phase of ARDS. Clin. Find. Eff. Corticosteroids. Chest 1991, 100, 943–952. [Google Scholar] [CrossRef]
- Meduri, G.U.; Chinn, A.J.; Leeper, K.V.; Wunderink, R.G.; Tolley, E.; Winer-Muram, H.T.; Khare, V.; Eltorky, M. Corticosteroid rescue treatment of progressive fibroproliferation in late ARDS: Patterns Response Predict. Outcome. Chest 1994, 105, 1516–1527. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Headley, S.; Tolley, E.; Shelby, M.; Stentz, F.; Postlethwaite, A. Plasma and BAL cytokine response to corticosteroid rescue treatment in late ARDS. Chest 1995, 108, 1315–1325. [Google Scholar] [CrossRef]
- Hakkinen, P.J.; Schmoyer, R.L.; Witschi, H.P. Potentiation of butylated-hydroxytoluene-induced acute lung damage by oxygen. Effects of prednisolone and indomethacin. Am. Rev. Respir. Dis. 1983, 128, 648–651. [Google Scholar] [PubMed]
- Hesterberg, T.W.; Last, J.A. Ozone-induced acute pulmonary fibrosis in rats. Prevention of increased rates of collagen synthesis by methylprednisolone. Am. Rev. Respir. Dis. 1981, 123, 47–52. [Google Scholar] [PubMed]
- Kehrer, J.P.; Klein-Szanto, A.J.; Sorensen, E.M.; Pearlman, R.; Rosner, M.H. Enhanced acute lung damage following corticosteroid treatment. Am. Rev. Respir. Dis. 1984, 130, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, K.; Ehrchen, J.; Tenbrock, K.; Ahlmann, M.; Kneidl, J.; Viemann, D.; Roth, J. Glucocorticoids promote survival of anti-inflammatory macrophages via stimulation of adenosine receptor A3. Blood 2010, 116, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrchen, J.; Steinmüller, L.; Barczyk, K.; Tenbrock, K.; Nacken, W.; Eisenacher, M.; Nordhues, U.; Sorg, C.; Sunderkötter, C.; Roth, J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood 2007, 109, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Liberman, A.C.; Budziñski, M.L.; Sokn, C.; Gobbini, R.P.; Steininger, A.; Arzt, E. Regulatory and Mechanistic Actions of Glucocorticoids on T and Inflammatory Cells. Front. Endocrinol. 2018, 9, 235. [Google Scholar] [CrossRef]
- Besedovsky, L.; Born, J.; Lange, T. Endogenous glucocorticoid receptor signaling drives rhythmic changes in human T-cell subset numbers and the expression of the chemokine receptor CXCR4. FASEB J. 2014, 28, 67–75. [Google Scholar] [CrossRef]
- Bouillet, P.; Metcalf, D.; Huang, D.C.; Tarlinton, D.M.; Kay, T.W.; Köntgen, F.; Adams, J.M.; Strasser, A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 1999, 286, 1735–1738. [Google Scholar] [CrossRef]
- Purton, J.F.; Monk, J.A.; Liddicoat, D.R.; Kyparissoudis, K.; Sakkal, S.; Richardson, S.J.; Godfrey, D.I.; Cole, T.J. Expression of the glucocorticoid receptor from the 1A promoter correlates with T lymphocyte sensitivity to glucocorticoid-induced cell death. J. Immunol. 2004, 173, 3816–3824. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 2014, 7, a016345. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielińska, K.A.; Van Moortel, L.; Opdenakker, G.; De Bosscher, K.; Van den Steen, P.E. Endothelial Response to Glucocorticoids in Inflammatory Diseases. Front. Immunol. 2016, 7, 592. [Google Scholar] [CrossRef] [Green Version]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef]
- Gavins, F.N.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef] [Green Version]
- Simoncini, T.; Maffei, S.; Basta, G.; Barsacchi, G.; Genazzani, A.R.; Liao, J.K.; De Caterina, R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res. 2000, 87, 19–25. [Google Scholar] [CrossRef]
- Gelati, M.; Corsini, E.; De Rossi, M.; Masini, L.; Bernardi, G.; Massa, G.; Boiardi, A.; Salmaggi, A. Methylprednisolone acts on peripheral blood mononuclear cells and endothelium in inhibiting migration phenomena in patients with multiple sclerosis. Arch. Neurol. 2002, 59, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, L.C.; de Kruif, M.D.; Giebelen, I.A.; van Zoelen, M.A.; van’t Veer, C.; van der Poll, T. Differential dose-dependent effects of prednisolone on shedding of endothelial adhesion molecules during human endotoxemia. Immunol. Lett. 2008, 121, 93–96. [Google Scholar] [CrossRef]
- Förster, C.; Kahles, T.; Kietz, S.; Drenckhahn, D. Dexamethasone induces the expression of metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular endothelial cell line cEND. J. Physiol. 2007, 580, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Kashiwamura, Y.; Sano, Y.; Abe, M.; Shimizu, F.; Haruki, H.; Maeda, T.; Kawai, M.; Kanda, T. Hydrocortisone enhances the function of the blood-nerve barrier through the up-regulation of claudin-5. Neurochem. Res. 2011, 36, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Limbourg, F.P.; Huang, Z.; Plumier, J.C.; Simoncini, T.; Fujioka, M.; Tuckermann, J.; Schütz, G.; Moskowitz, M.A.; Liao, J.K. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J. Clin. Investig. 2002, 110, 1729–1738. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, S.; Yang, Y.; Yao, J.Q.; Tang, W.F.; Lyon, C.J.; Hu, T.Y.; Wan, M.H. Extracellular vesicles in the pathogenesis and treatment of acute lung injury. Mil. Med. Res. 2022, 9, 61. [Google Scholar] [CrossRef]
- Shah, J.; Rana, S.S. Acute respiratory distress syndrome in acute pancreatitis. Indian. J. Gastroenterol. 2020, 39, 123–132. [Google Scholar] [CrossRef]
- Zielińska, K.A.; de Cauwer, L.; Knoops, S.; Van der Molen, K.; Sneyers, A.; Thommis, J.; De Souza, J.B.; Opdenakker, G.; De Bosscher, K.; Van den Steen, P.E. Plasmodium berghei NK65 in Combination with IFN-γ Induces Endothelial Glucocorticoid Resistance via Sustained Activation of p38 and JNK. Front. Immunol. 2017, 8, 1199. [Google Scholar] [CrossRef] [Green Version]
- Dschietzig, T.; Richter, C.; Pfannenschmidt, G.; Bartsch, C.; Laule, M.; Baumann, G.; Stangl, K. Dexamethasone inhibits stimulation of pulmonary endothelins by proinflammatory cytokines: Possible involvement of a nuclear factor kappa B dependent mechanism. Intensive Care Med. 2001, 27, 751–756. [Google Scholar] [CrossRef]
- Singh, P.; Garg, P.K. Pathophysiological mechanisms in acute pancreatitis: Current understanding. Indian. J. Gastroenterol. 2016, 35, 153–166. [Google Scholar] [CrossRef]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Mallédant, Y.; Malbrain, M.L.; Reuter, D.A. What’s new in the management of severe acute pancreatitis. Intensive Care Med. 2015, 41, 1957–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.J.; Chen, J.; Phillips, A.R.; Windsor, J.A.; Petrov, M.S. Predictors of severe and critical acute pancreatitis: A systematic review. Dig. Liver Dis. 2014, 46, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Semler, M.W.; Self, W.H.; Wanderer, J.P.; Ehrenfeld, J.M.; Wang, L.; Byrne, D.W.; Stollings, J.L.; Kumar, A.B.; Hughes, C.G.; Hernandez, A.; et al. Balanced Crystalloids versus Saline in Critically Ill Adults. N. Engl. J. Med. 2018, 378, 829–839. [Google Scholar] [CrossRef]
- Brown, L.A.; Hore, T.A.; Phillips, A.R.; Windsor, J.A.; Petrov, M.S. A systematic review of the extra-pancreatic infectious complications in acute pancreatitis. Pancreatology 2014, 14, 436–443. [Google Scholar] [CrossRef]
- Lim, C.L.; Lee, W.; Liew, Y.X.; Tang, S.S.; Chlebicki, M.P.; Kwa, A.L. Role of antibiotic prophylaxis in necrotizing pancreatitis: A meta-analysis. J. Gastrointest. Surg. 2015, 19, 480–491. [Google Scholar] [CrossRef]
- De Waele, E.; Malbrain, M.; Spapen, H.D. How to deal with severe acute pancreatitis in the critically ill. Curr. Opin. Crit. Care 2019, 25, 150–156. [Google Scholar] [CrossRef]
- Qadir, N.; Bartz, R.R.; Cooter, M.L.; Hough, C.L.; Lanspa, M.J.; Banner-Goodspeed, V.M.; Chen, J.T.; Giovanni, S.; Gomaa, D.; Sjoding, M.W.; et al. Variation in Early Management Practices in Moderate-to-Severe ARDS in the United States: The Severe ARDS: Generating Evidence Study. Chest 2021, 160, 1304–1315. [Google Scholar] [CrossRef]
- Shimosegawa, T. Are glucocorticoids really useful for the treatment of acute pancreatitis. J. Gastroenterol. 2002, 37, 580–581. [Google Scholar] [CrossRef]
- Stephenson, H.E., Jr.; Pfeffer, R.B.; Saypol, G.M. Acute hemorrhagic pancreatitis: Report of a case with cortisone treatment. A.M.A. Arch. Surg. 1952, 65, 307–308. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, Z.; Liang, H. Glucocorticoids in acute pancreatitis: A propensity score matching analysis. BMC Gastroenterol. 2021, 21, 331. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Liu, Z.M.; Wang, S.J.; Zhao, S.J.; Zhang, D.; Chen, Y.; Wang, Y.S. Corticosteroid therapy for severe acute pancreatitis: A meta-analysis of randomized, controlled trials. Int. J. Clin. Exp. Pathol. 2015, 8, 7654–7660. [Google Scholar] [PubMed]
- Pastores, S.M.; Annane, D.; Rochwerg, B.; Corticosteroid Guideline Task Force of SCCM and ESICM. Guidelines for the diagnosis and management of critical illness-related corticosteroid insufficiency (CIRCI) in critically ill patients (Part II): Society of Critical Care Medicine (SCCM) and European Society of Intensive Care Medicine (ESICM) 2017. Intensive Care Med. 2018, 44, 474–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keel, J.B.; Hauser, M.; Stocker, R.; Baumann, P.C.; Speich, R. Established acute respiratory distress syndrome: Benefit of corticosteroid rescue therapy. Respiration 1998, 65, 258–264. [Google Scholar] [CrossRef]
- Meduri, G.U.; Golden, E.; Freire, A.X.; Taylor, E.; Zaman, M.; Carson, S.J.; Gibson, M.; Umberger, R. Methylprednisolone infusion in early severe ARDS: Results of a randomized controlled trial. Chest 2007, 131, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Meduri, G.U.; Headley, A.S.; Golden, E.; Carson, S.J.; Umberger, R.A.; Kelso, T.; Tolley, E.A. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: A randomized controlled trial. JAMA 1998, 280, 159–165. [Google Scholar] [CrossRef]
- Steinberg, K.P.; Hudson, L.D.; Goodman, R.B.; Hough, C.L.; Lanken, P.N.; Hyzy, R.; Thompson, B.T.; Ancukiewicz, M.; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N. Engl. J. Med. 2006, 354, 1671–1684. [Google Scholar]
- Umberto Meduri, G.; Bell, W.; Sinclair, S.; Annane, D. Pathophysiology of acute respiratory distress syndrome: Glucocorticoid receptor-mediated regulation of inflammation and response to prolonged glucocorticoid treatment. Presse Méd. 2011, 40, e543–e560. [Google Scholar] [CrossRef]
- Meduri, G.U.; Yates, C.R. Systemic inflammation-associated glucocorticoid resistance and outcome of ARDS. Ann. N. Y. Acad. Sci. 2004, 1024, 24–53. [Google Scholar] [CrossRef]
- Balani, A.R.; Grendell, J.H. Drug-induced pancreatitis: Incidence, management and prevention. Drug Saf. 2008, 31, 823–837. [Google Scholar] [CrossRef]
- Nango, D.; Hirose, Y.; Goto, M.; Echizen, H. Analysis of the Association of Administration of various glucocorticoids with development of acute pancreatitis using US Food and Drug Administration adverse event reporting system (FAERS). J. Pharm. Health Care Sci. 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadr-Azodi, O.; Mattsson, F.; Bexlius, T.S.; Lindblad, M.; Lagergren, J.; Ljung, R. Association of oral glucocorticoid use with an increased risk of acute pancreatitis: A population-based nested case-control study. JAMA Intern. Med. 2013, 173, 444–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, Y.; Ogasa, S.; Izaki, S.; Kitamura, K. Corticosteroid-induced pancreatitis in patients with autoimmune bullous disease: Case report and prospective study. Dermatology 1999, 198, 304–306. [Google Scholar] [CrossRef]
- Dale, D.C.; Petersdorf, R.G. Corticosteroids and infectious diseases. Med. Clin. N. Am. 1973, 57, 1277–1287. [Google Scholar] [CrossRef]
- MacGregor, R.R.; Sheagren, J.N.; Lipsett, M.B.; Wolff, S.M. Alternate-day prednisone therapy. Evaluation of delayed hypersensitivity responses, control of disease and steroid side effects. N. Engl. J. Med. 1969, 280, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S.; Kontoyiannis, D.P. Glucocorticoids and invasive fungal infections. Lancet 2003, 362, 1828–1838. [Google Scholar] [CrossRef]
- Conn, H.O.; Blitzer, B.L. Nonassociation of adrenocorticosteroid therapy and peptic ulcer. N. Engl. J. Med. 1976, 294, 473–479. [Google Scholar] [CrossRef]
- Messer, J.; Reitman, D.; Sacks, H.S.; Smith, H., Jr.; Chalmers, T.C. Association of adrenocorticosteroid therapy and peptic-ulcer disease. N. Engl. J. Med. 1983, 309, 21–24. [Google Scholar] [CrossRef]
- Luo, J.C.; Shin, V.Y.; Liu, E.S.; Ye, Y.N.; Wu, W.K.; So, W.H.; Chang, F.Y.; Cho, C.H. Dexamethasone delays ulcer healing by inhibition of angiogenesis in rat stomachs. Eur. J. Pharmacol. 2004, 485, 275–281. [Google Scholar] [CrossRef]
- Narum, S.; Westergren, T.; Klemp, M. Corticosteroids and risk of gastrointestinal bleeding: A systematic review and meta-analysis. BMJ Open 2014, 4, e004587. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Greenspan, A.; Gershwin, M.E. The pathogenesis, diagnosis and clinical manifestations of steroid-induced osteonecrosis. J. Autoimmun. 2020, 110, 102460. [Google Scholar] [CrossRef] [PubMed]
- Koo, K.H.; Kim, R.; Kim, Y.S.; Ahn, I.O.; Cho, S.H.; Song, H.R.; Park, Y.S.; Kim, H.; Wang, G.J. Risk period for developing osteonecrosis of the femoral head in patients on steroid treatment. Clin. Rheumatol. 2002, 21, 299–303. [Google Scholar] [CrossRef]
- Campbell, K.L.; Latimer, K.S. Transient diabetes mellitus associated with prednisone therapy in a dog. J. Am. Vet. Med. Assoc. 1984, 185, 299–301. [Google Scholar] [PubMed]
- Jeffers, J.G.; Shanley, K.J.; Schick, R.O. Diabetes mellitus induced in a dog after administration of corticosteroids and methylprednisolone pulse therapy. J. Am. Vet. Med. Assoc. 1991, 199, 77–80. [Google Scholar]
- Yubero, S.; Manso, M.A.; Ramudo, L.; Vicente, S.; De Dios, I. Dexamethasone down-regulates the inflammatory mediators but fails to reduce the tissue injury in the lung of acute pancreatitis rat models. Pulm. Pharmacol. Ther. 2012, 25, 319–324. [Google Scholar] [CrossRef]
- Von Mässenhausen, A.; Zamora Gonzalez, N.; Maremonti, F.; Belavgeni, A.; Tonnus, W.; Meyer, C.; Beer, K.; Hannani, M.T.; Lau, A.; Peitzsch, M.; et al. Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci. Adv. 2022, 8, eabl8920. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Sébille, V.; Bellissant, E.; Ger-Inf-05 Study Group. Effect of low doses of corticosteroids in septic shock patients with or without early acute respiratory distress syndrome. Crit. Care Med. 2006, 34, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Sabry, N.A.; Omar, E.E.-D. Corticosteroids and ICU Course of Community Acquired Pneumonia in Egyptian Settings. Pharmacol. Pharm. 2011, 2, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Confalonieri, M.; Urbino, R.; Potena, A.; Piattella, M.; Parigi, P.; Puccio, G.; Della Porta, R.; Giorgio, C.; Blasi, F.; Umberger, R.; et al. Hydrocortisone infusion for severe community-acquired pneumonia: A preliminary randomized study. Am. J. Respir. Crit. Care Med. 2005, 171, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Marik, P.; Kraus, P.; Sribante, J.; Havlik, I.; Lipman, J.; Johnson, D.W. Hydrocortisone and tumor necrosis factor in severe community-acquired pneumonia. A randomized controlled study. Chest 1993, 104, 389–392. [Google Scholar] [CrossRef]
- Tongyoo, S.; Permpikul, C.; Mongkolpun, W.; Vattanavanit, V.; Udompanturak, S.; Kocak, M.; Meduri, G.U. Hydrocortisone treatment in early sepsis-associated acute respiratory distress syndrome: Results of a randomized controlled trial. Crit. Care 2016, 20, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, G.R.; Luce, J.M.; Sprung, C.L.; Rinaldo, J.E.; Tate, R.M.; Sibbald, W.J.; Kariman, K.; Higgins, S.; Bradley, R.; Metz, C.A.; et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N. Engl. J. Med. 1987, 317, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Lee, J.M.; Kim, M.S.; Kim, H.Y.; Hwangbo, B.; Zo, J.I. Low-dose steroid therapy at an early phase of postoperative acute respiratory distress syndrome. Ann. Thorac. Surg. 2005, 79, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Varpula, T.; Rintala, E.; Takkunen, O.; Valtonen, V.; Pettilä, V. Late steroid therapy in primary acute lung injury. Intensive Care Med. 2000, 26, 526–531. [Google Scholar] [CrossRef]
- Weigelt, J.A.; Norcross, J.F.; Borman, K.R.; Snyder, W.H. Early steroid therapy for respiratory failure. Arch. Surg. 1985, 120, 536–540. [Google Scholar] [CrossRef]
- Wan, M.-H.; Li, J.; Gong, H.-L.; Xue, P.; Zhu, L.; Chen, G.-Y.; Xia, Q.; Wen-Fu, T. Clinical observation on the effect of dexamethasone and Chinese herbal decoction for purgation in severe acute pancreatitis patients. Chin. J. Integr. Med. 2011, 17, 141–145. [Google Scholar] [CrossRef]
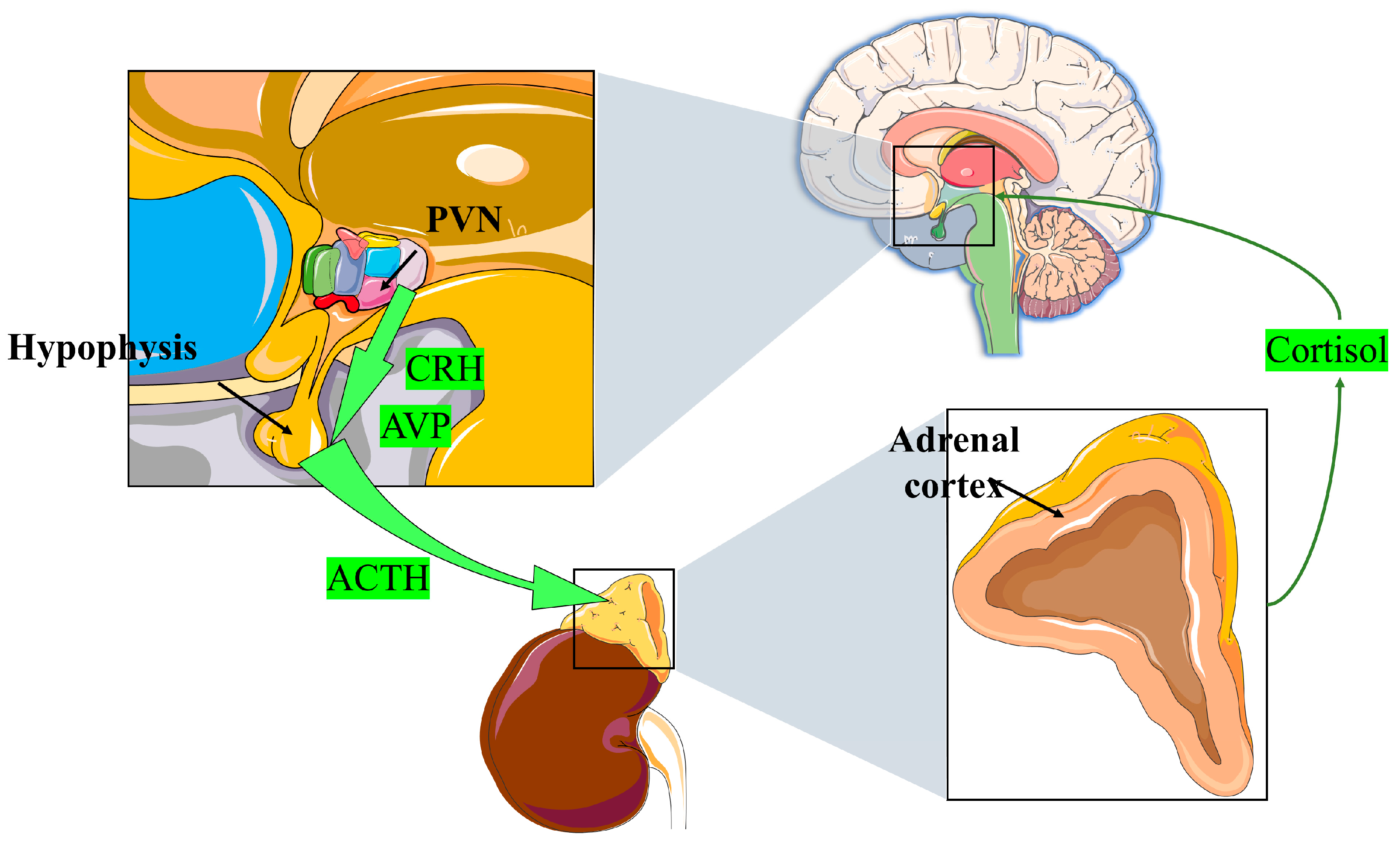
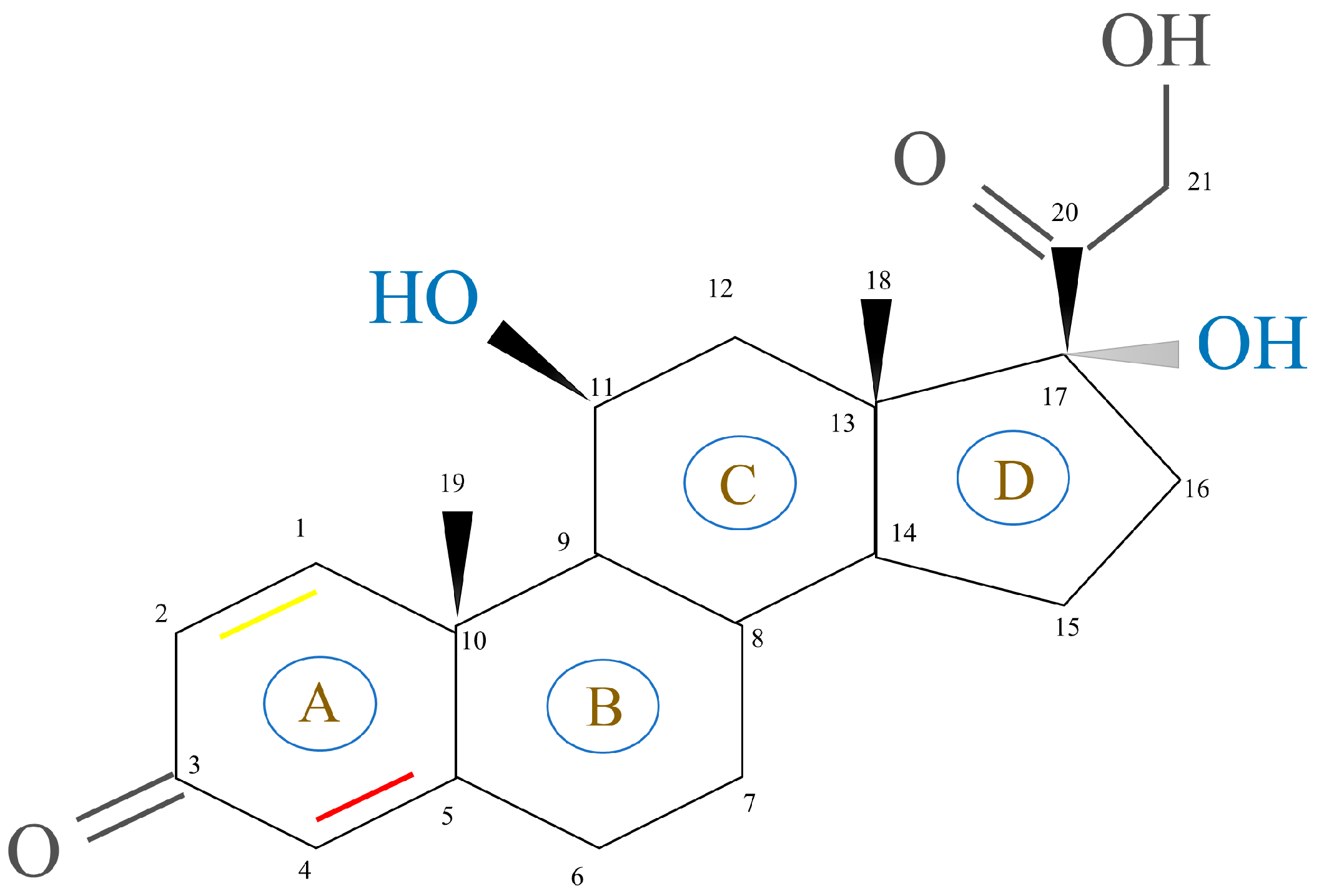
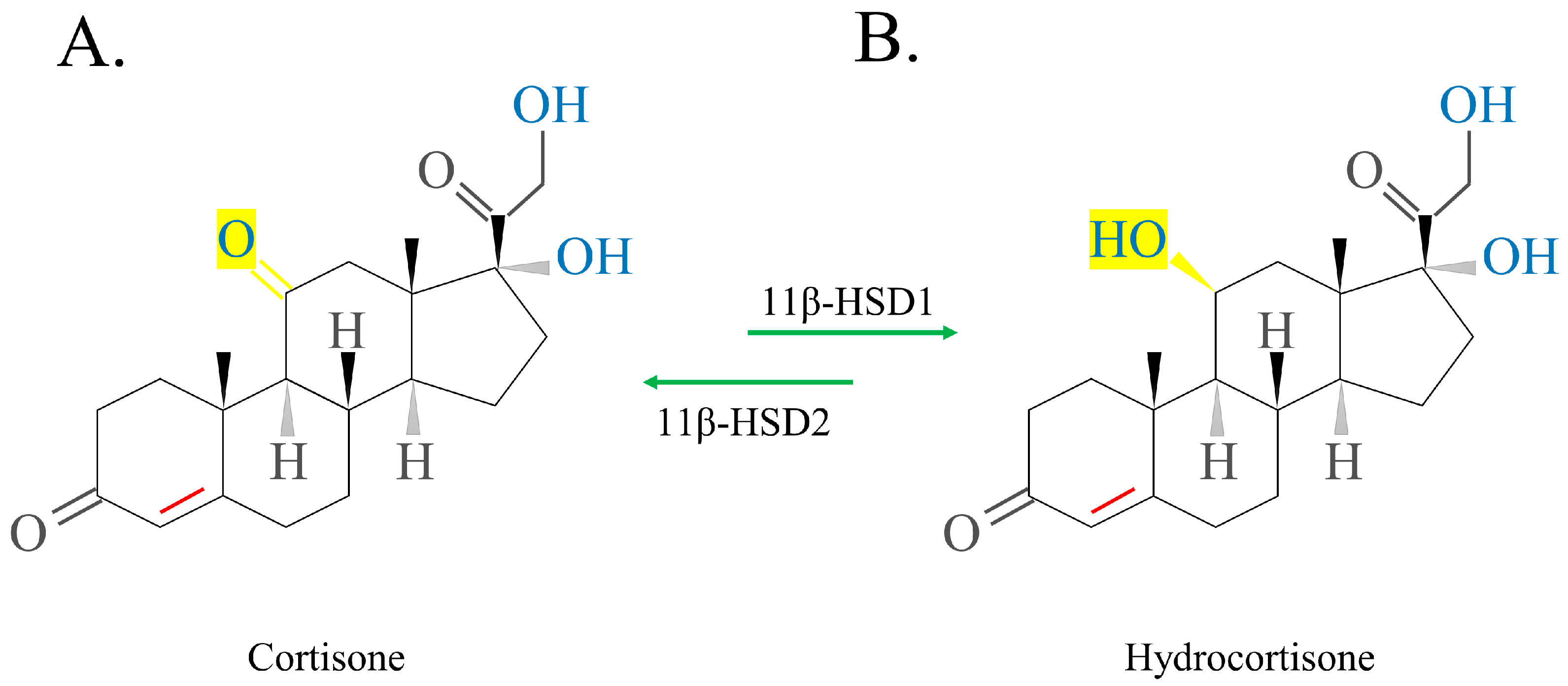
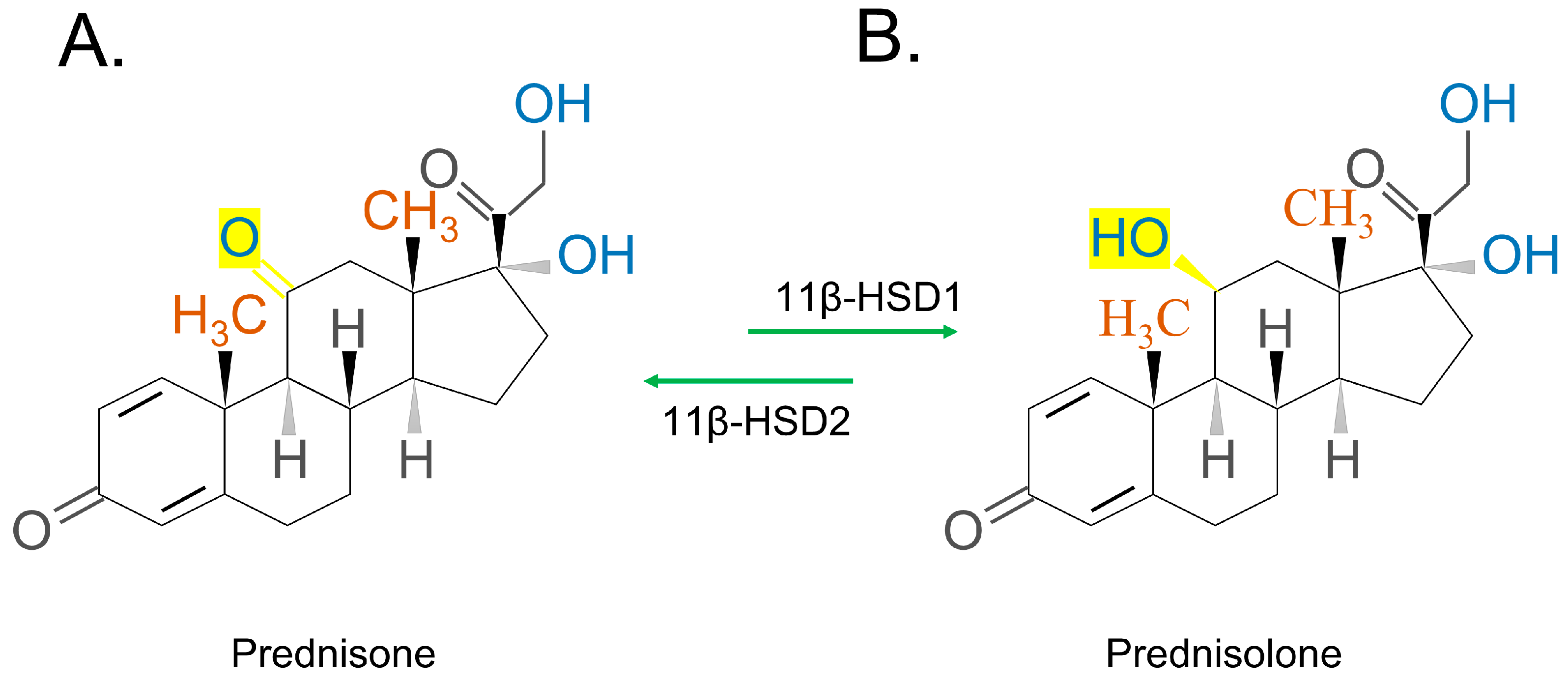

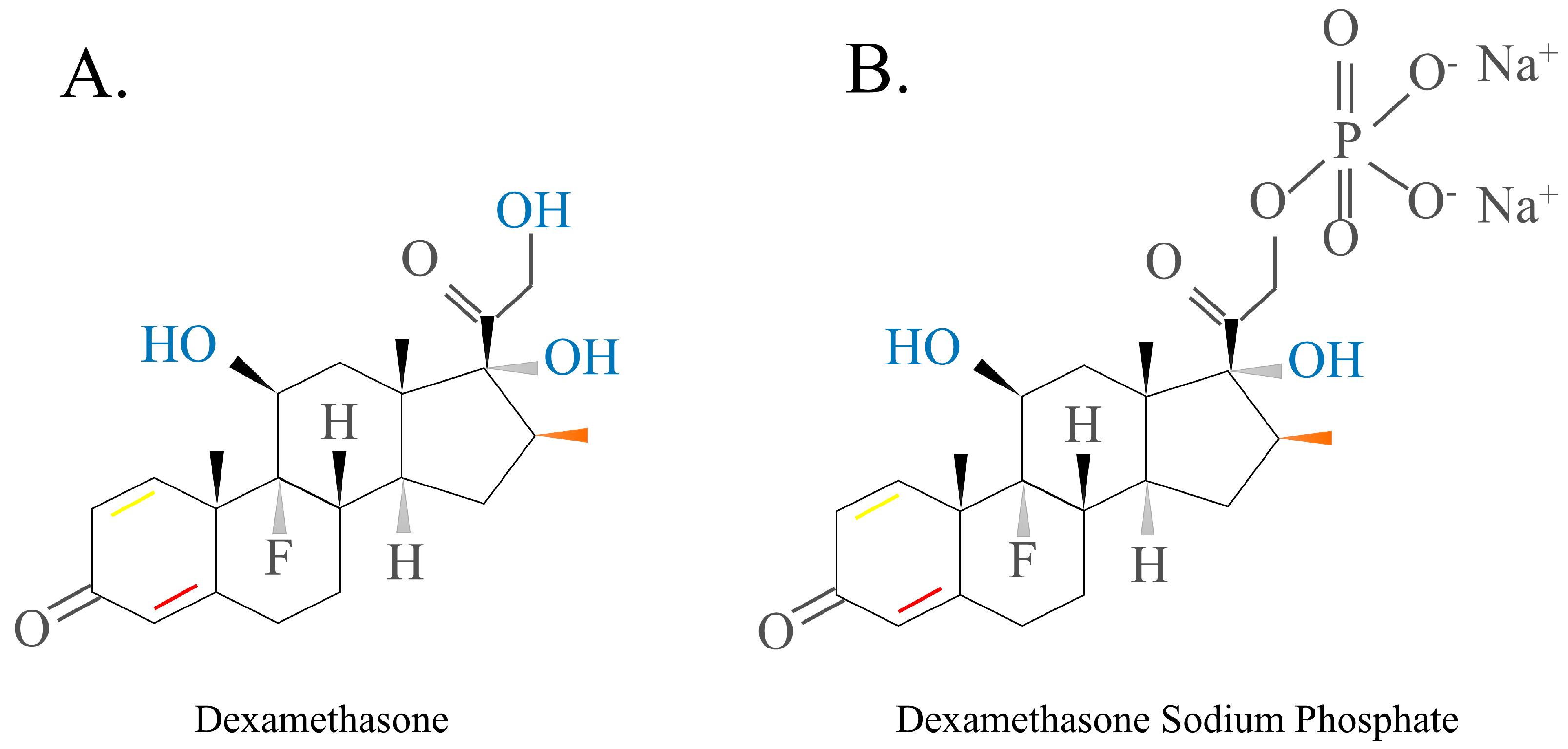

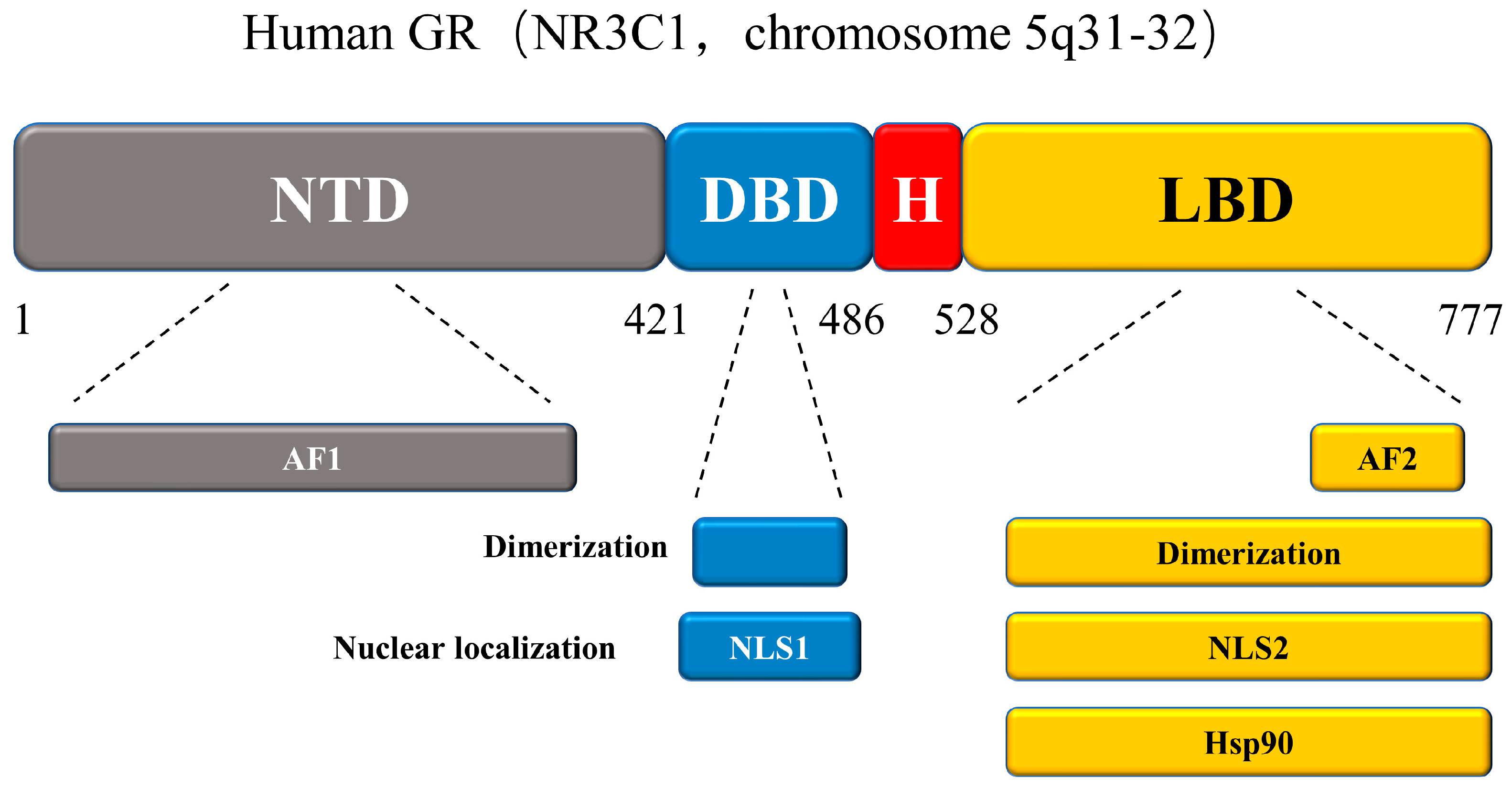
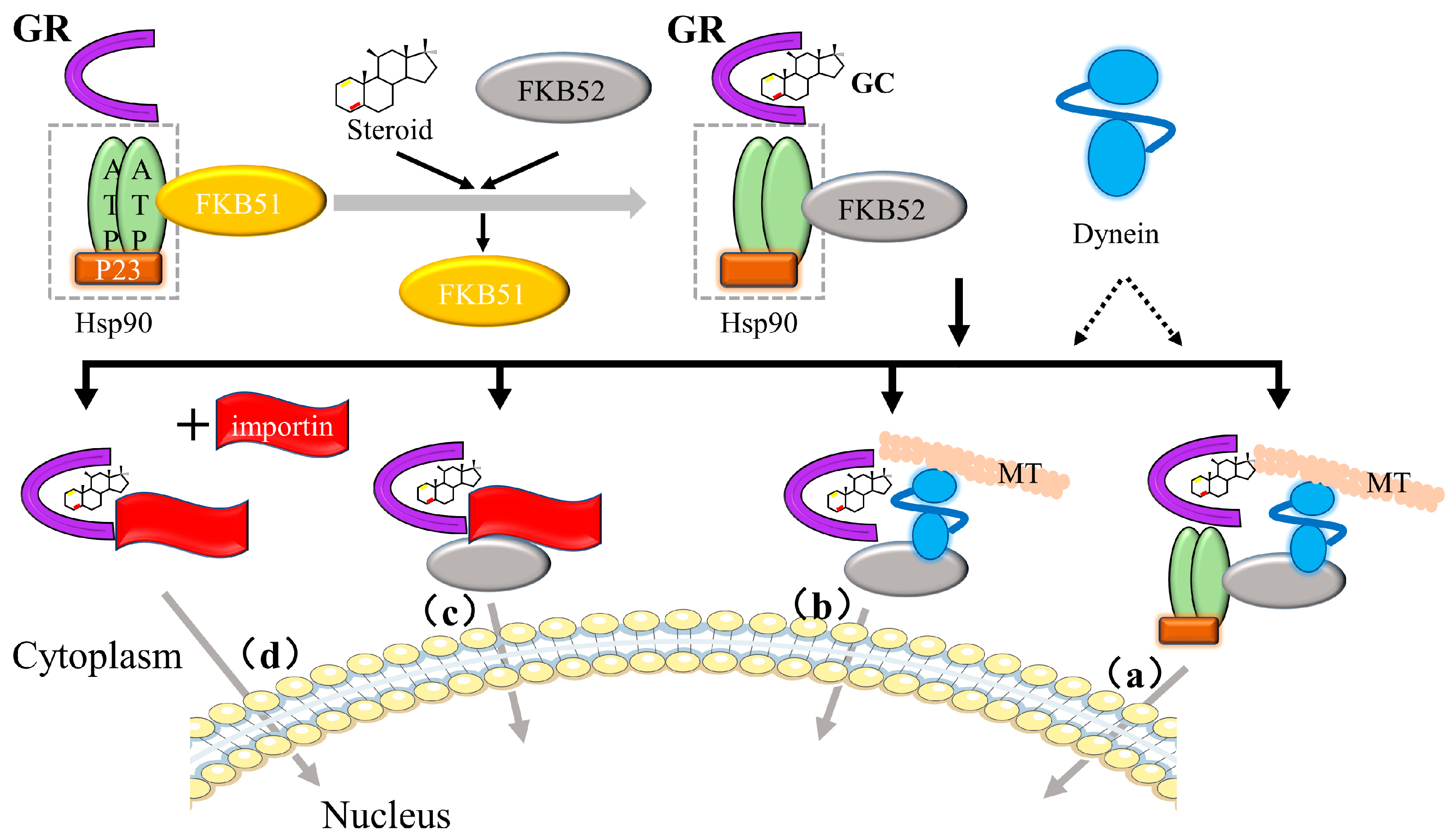
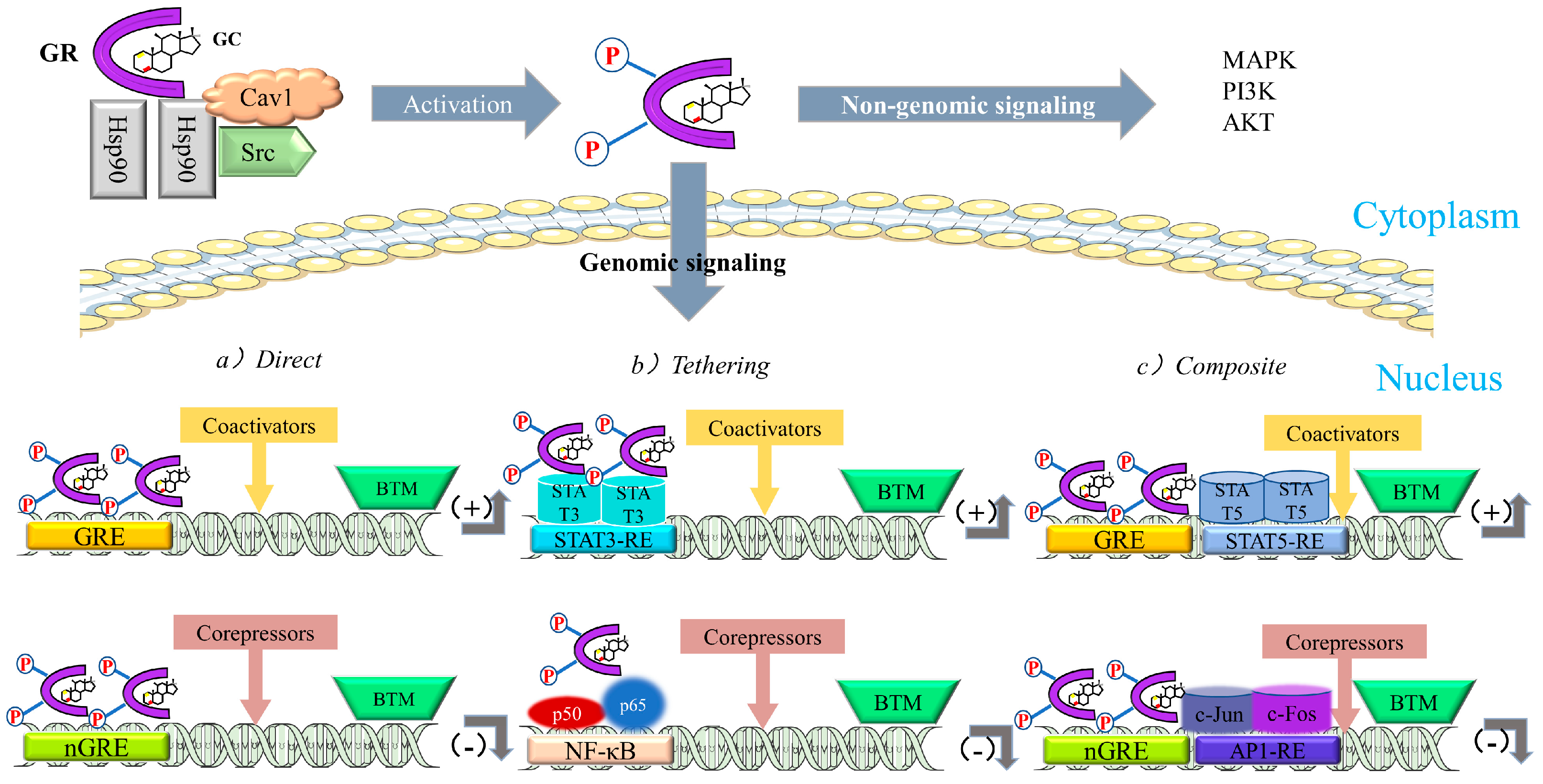

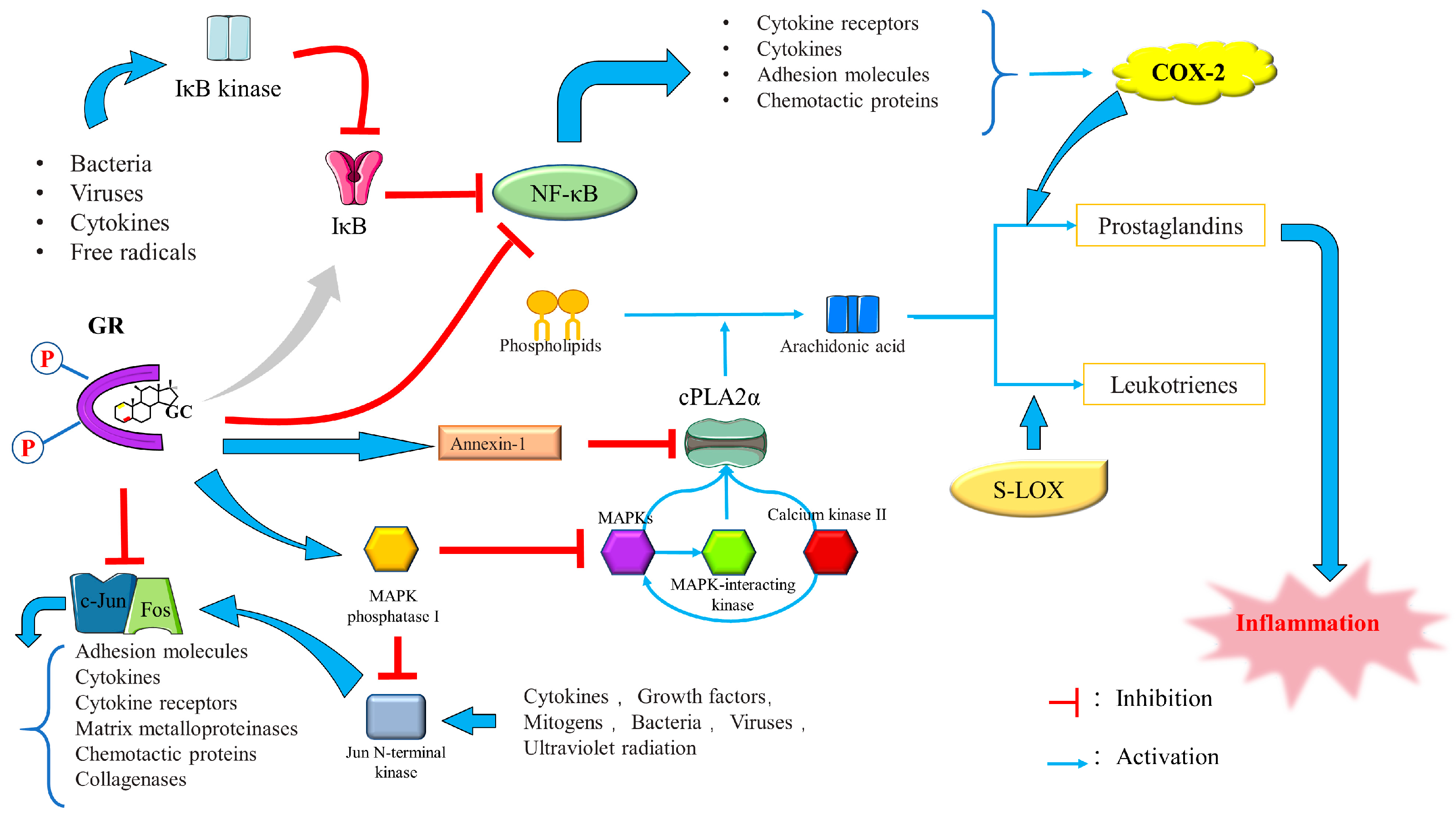
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ge, P.; Liu, J.; Luo, Y.; Guo, H.; Zhang, G.; Xu, C.; Chen, H. Glucocorticoid Treatment in Acute Respiratory Distress Syndrome: An Overview on Mechanistic Insights and Clinical Benefit. Int. J. Mol. Sci. 2023, 24, 12138. https://doi.org/10.3390/ijms241512138
Zhang J, Ge P, Liu J, Luo Y, Guo H, Zhang G, Xu C, Chen H. Glucocorticoid Treatment in Acute Respiratory Distress Syndrome: An Overview on Mechanistic Insights and Clinical Benefit. International Journal of Molecular Sciences. 2023; 24(15):12138. https://doi.org/10.3390/ijms241512138
Chicago/Turabian StyleZhang, Jinquan, Peng Ge, Jie Liu, Yalan Luo, Haoya Guo, Guixin Zhang, Caiming Xu, and Hailong Chen. 2023. "Glucocorticoid Treatment in Acute Respiratory Distress Syndrome: An Overview on Mechanistic Insights and Clinical Benefit" International Journal of Molecular Sciences 24, no. 15: 12138. https://doi.org/10.3390/ijms241512138