
Calcitonin Gene-Related Peptide-Mediated Trigeminal Ganglionitis: The Biomolecular Link between Temporomandibular Disorders and Chronic Headaches

Abstract
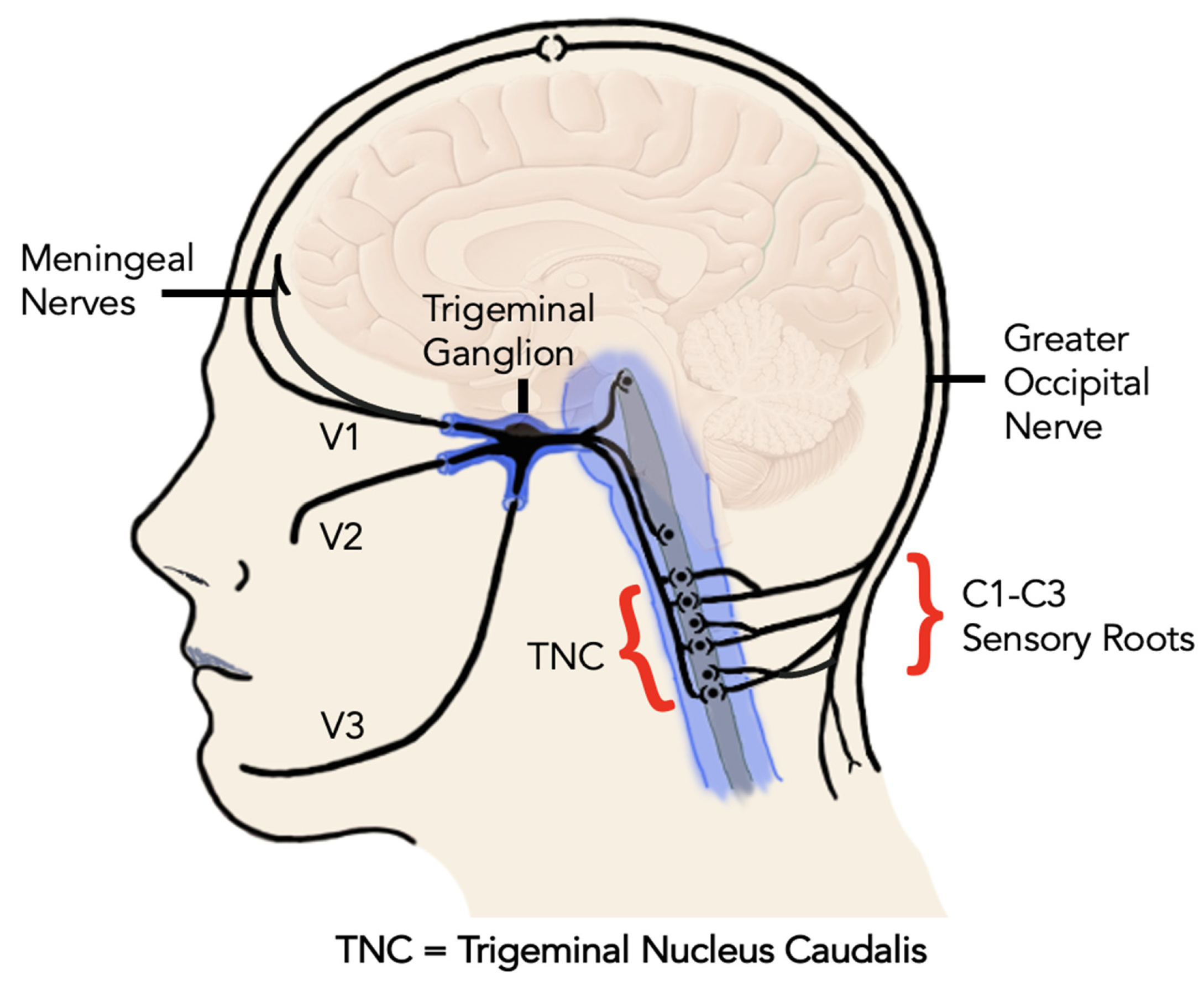
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Study Selection Criteria
3. CGRP in Peripheral and Central Sensitization
- Peripheral sensitization. Peripheral injury of the muscles, joints, or nerves of the jaw and head triggers pain signals in the trigeminal nerve from the primary afferent fibers (mostly C fiber and A delta fibers). Local tissue inflammation releases cytokines and pro-inflammatory mediators, including CGRP, that perpetuate and increase the pain response. Peripheral sensitization lowers the depolarization threshold, so that normal stimulation is now perceived as painful (primary allodynia) and painful stimuli result in higher pain perception (primary hyperalgesia or “hyperalgesic priming”) [16,17].
- Central sensitization. Sustained peripheral pain signaling leads to central sensitization, characterized by increased excitability of central pain pathways [18] https://www.iasp-pain.org/resources/terminology/ (Accessed on 25 February 2023) [18,19]. At first, this sensitization is activity-dependent and consists primarily of lowered depolarization thresholds. This characterizes the phase of acute pain. However, if it persists for a longer period of time (i.e., beyond the normal healing process), it evolves into an activity-independent phenomenon through neuroplastic adaptation [14]. In this scenario, the CGRP released in the trigeminal ganglion engages with adjacent neurons and satellite glial cells, causing the continuation of peripheral sensitization and facilitating central sensitization of the second-order neurons [13]. This identifies a shift to a chronic pain phase. Central sensitization is the physiological hallmark of chronic pain syndromes and is responsible for the clinical symptoms of secondary hyperalgesia (defined as the increased pain response derived from a normally painful stimulus (Terminology | International Association for the Study of Pain. International Association for the Study of Pain (IASP). https://www.iasp-pain.org/resources/terminology/ (Accessed on 25 February 2023) [18]) and secondary allodynia (defined as pain response derived from a stimulus that is not normally perceived as painful [19].
- Spinal cord, where CGRP is released from primary sensory neurons in the dorsal horn of the spinal cord and cerebral gray matter, where it contributes to pain transmission and modulation [21];
- Brainstem: CGRP-containing fibers and terminals have been identified in various brainstem nuclei involved in pain processing, including the periaqueductal gray (PAG) and the nucleus tractus solitarius (NTS);
- Hypothalamus: CGRP has been detected in certain hypothalamic nuclei, such as the paraventricular nucleus (PVN) and the supraoptic nucleus (SON), involved in the regulation of autonomic functions and pain modulation [21];
- Thalamus: neurons expressing CGRP in the parvocellular sub-parafascicular nucleus have been observed in the thalamus [25].
4. CGRP and Migraine
- Ergotamine derivatives and triptans, drugs approved for the acute treatment of migraine [38], mainly stimulate 5-HT1B/1D receptors. As 5-HT1B receptor is localized on smooth muscle cells of cerebral, meningeal and coronary arteries, and 5-HT1D is mainly expressed in the trigeminal ganglion, these drugs result in a strong vasoconstriction of the cranial arteries [39]. They also indirectly act on decreasing the release of CGRP, thus reducing trigeminal activation and vasodilation [35].
- Ditans, 5-HT1F receptor agonists (lasmiditan) are newly Food and Drug Administration (FDA)-approved drugs for the acute treatment of migraine [40]. 5- HT1F receptors are located on terminals and cell bodies of the trigeminal ganglion neurons, acting at the peripheral nervous system and central nervous system. It can modulate CGRP from trigeminal ganglion neurons by potentially blocking its release and inhibiting the development of central sensitization [40]. Contrary to the effect of ergotamine derivates and triptans, activation of 5- HT1F does not induce vasodilation but rather causes vasoconstriction [40].
- CGRP receptors are localized on smooth muscles cells of meningeal and cerebral blood vessels. As a result, the release of CGRP by the activated meningeal C-fibers causes blood vessels to dilate [37]. Direct blockade of CGRP signaling with gepants [41], and with monoclonal antibodies directed against the molecule or its receptor attenuated the cutaneous mechanical hypersensitivity [42] and nitroglycerin-induced trigeminal hyperalgesia in animal models of migraine-like pain [43]. They have been FDA-approved as an effective treatment in preventive migraine [44]. Although the exact mechanism and site of action of CGRP in pain are still unclear and many mechanisms of action have been proposed [45], the meningeal blood vessels and their vasodilation are a primary target to prevent or inhibit pain signals [23].
- Topiramate inhibits nitric oxide and proton mediated CGRP secretion in a time- and concentration-dependent fashion from sensory trigeminal neurons [46].
- Botulinum toxin-A (BoNT) at doses between 150 Units and 195 Units, repeated every 12 weeks, is listed among the FDA-approved therapies for prevention of chronic migraine [38]. Among the several hypotheses on mechanism of action, one of these supports that BoNT attenuates the release of local transmitters such as CGRP from trigeminal neurons [47]. This further supports the pivotal role of CGRP reduction to its mechanism of action [48].
5. CGRP and TMDs
- CGRP receptors are widely distributed through the trigeminovascular system [71].
- The inflammatory response from peripheral injury stimulates the expression of CGRP in the trigeminal ganglion and central relay centers [13].
- Biomolecular CRGP release in the trigeminal ganglion stimulates the release of pro-inflammatory mediators via supporting cells in a paracrine fashion [72].
- The resulting “inflammatory soup” in the ganglion (a sterile ganglionitis) is permissive of cross-excitation of all branches of the trigeminal nerve [73].
6. CGRP, Traumatic Headaches and TMD
- Direct jaw trauma;
- Sports injuries;
- Motor vehicle accidents;
- Whiplash associated injuries;
- Hyperextension injuries;
- Strain from repetitive or continuous muscle activation;
- Bruxism and other parafunctional behaviors.
7. Treatment Considerations
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Okeson, J.P. Bell’s Oral and Facial Pain, 7th ed; CBS: New York, NY, USA, 2014. [Google Scholar]
- Facial Pain. Available online: https://www.nidcr.nih.gov/research/data-statistics/facial-pain (accessed on 24 February 2023).
- Orofacial Pain: Guidelines for Assessment, Diagnosis, and Management—NLM Catalog—NCBI. Available online: https://www.ncbi.nlm.nih.gov/nlmcatalog/101603146 (accessed on 25 February 2023).
- Woldeamanuel, Y.W.; Cowan, R.P. Migraine affects 1 in 10 people worldwide featuring recent rise: A systematic review and meta-analysis of community-based studies involving 6 million participants. J. Neurol. Sci. 2017, 372, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Scripter, C. Headache: Tension-Type Headache. FP Essent. 2018, 473, 17–20. [Google Scholar]
- Schiffman, E.; Ohrbach, R.; Truelove, E.; Look, J.; Anderson, G.; Goulet, J.-P.; List, T.; Svensson, P.; Gonzalez, Y.; Lobbezoo, F.; et al. Diagnostic Criteria for Temporomandibular Disorders (DC/TMD) for Clinical and Research Applications: Recommendations of the International RDC/TMD Consortium Network* and Orofacial Pain Special Interest Group. J. Oral. Facial Pain Headache 2014, 28, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Réus, J.C.; Polmann, H.; Souza, B.D.M.; Fores-Mir, C.; Goncalves, D.A.G.; de Queiroz, L.P.; Okeson, J.; Canto, G.D.L. Association between primary headaches and temporomandibular disorders: A systematic review and meta-analysis. J. Am. Dent. Assoc. 2022, 153, 120–131.e6. [Google Scholar] [CrossRef] [PubMed]
- Tchivileva, I.E.; Ohrbach, R.; Fillingim, R.B.; Greenspan, J.D.; Maixner, W.; Slade, G.D. Temporal change in headache and its contribution to risk of developing first-onset TMD in the OPPERA study. Pain 2017, 158, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meer, H.A.; Speksnijder, C.M.; Engelbert, R.H.H.; Lobbezoo, F.; Nijhuis-van der Sanden, M.W.G.; Visscher, C.M. The Association Between Headaches and Temporomandibular Disorders is Confounded by Bruxism and Somatic Symptoms. Clin. J. Pain 2017, 33, 835–843. [Google Scholar] [CrossRef]
- Gonçalves, D.A.G.; Bigal, M.E.; Jales, L.C.F.; Camparis, C.M.; Speciali, J.G. Headache and symptoms of temporomandibular disorder: An epidemiological study. Headache 2010, 50, 231–241. [Google Scholar] [CrossRef]
- Marklund, S.; Wiesinger, B.; Wänman, A. Reciprocal influence on the incidence of symptoms in trigeminally and spinally innervated areas. Eur. J. Pain Lond. Engl. 2010, 14, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Shinozaki, T.; Okada-Ogawa, A.; Matsukawa, Y.; Dezawa, K.; Nakaya, Y.; Chen, J.Y.; Noma, N.; Oka, S.; Iwata, K.; et al. Headache attributed to temporomandibular disorders and masticatory myofascial pain. J. Oral. Sci. 2016, 58, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, S.; Johnson, K.W.; Ossipov, M.H.; Aurora, S.K. CGRP and the Trigeminal System in Migraine. Headache 2019, 59, 659–681. [Google Scholar] [CrossRef] [Green Version]
- Durham, P.L. Diverse Physiological Roles of Calcitonin Gene-Related Peptide in Migraine Pathology: Modulation of Neuronal-Glial-Immune Cells to Promote Peripheral and Central Sensitization. Curr. Pain Headache Rep. 2016, 20, 48. [Google Scholar] [CrossRef] [Green Version]
- Cady, R.J.; Glenn, J.R.; Smith, K.M.; Durham, P.L. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol. Pain 2011, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Sessle, B.J. Peripheral and central mechanisms of orofacial inflammatory pain. Int. Rev. Neurobiol. 2011, 97, 179–206. [Google Scholar] [CrossRef]
- Reichling, D.B.; Levine, J.D. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009, 32, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terminology|International Association for the Study of Pain. International Association for the Study of Pain (IASP). Available online: https://www.iasp-pain.org/resources/terminology/ (accessed on 24 February 2023).
- Temporomandibular Disorders: Priorities for Research and Care. 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557989/ (accessed on 20 November 2021).
- Weston, C.; Winfiled, I.; Harris, M.; Hodgson, R.; Shah, A.; Dowell, S.J.; Mobarec, J.C.; Woodlock, D.A.; Reynolds, C.A.; Poyner, D.R.; et al. Receptor Activity-modifying Protein-directed G Protein Signaling Specificity for the Calcitonin Gene-related Peptide Family of Receptors. J. Biol. Chem. 2016, 291, 21925–22194. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, S.; Ossipov, M.H.; Johnson, K.W. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 2017, 158, 543–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalakoti, S.; Patil, V.V.; Damodaram, S.; Vause, C.V.; Langford, L.E.; Freeman, S.E.; Durham, P.L. Neuron-glia signaling in trigeminal ganglion: Implications for migraine pathology. Headache 2007, 47, 1008–1023; discussion 24–25. [Google Scholar] [CrossRef] [Green Version]
- Sangalli, L.; Brazzoli, S. Calcitonin Gene-Related Peptide (CGRP)-Targeted Treatments—New Therapeutic Technologies for Migraine. Future Pharmacol. 2023, 3, 117–131. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.M.; Flores, C.M.; Harding-Rose, C.A.; Goodis, H.E.; Hargreaves, K.M. Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from rat trigeminal ganglion: Evidence for intraganglionic neurotransmission. Pain 2001, 91, 219–226. [Google Scholar] [CrossRef]
- Kang, S.J.; Liu, S.; Ye, M.; Kim, D.I.; Pao, G.M.; Copits, B.A.; Roberts, B.Z.; Lee, K.F.; Bruchas, M.R.; Han, S. A central alarm system that gates multi-sensory innate threat cues to the amygdala. Cell. Rep. 2022, 40, 111222. [Google Scholar] [CrossRef]
- Mulderry, P.K.; Gathei, M.A.; Spokes, R.A.; Jones, P.M.; Pierson, A.M.; Hamid, Q.A.; Kanse, S.; Amara, S.G.; Burrin, J.M.; Legon, S.; et al. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 1988, 25, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef]
- Amir, R.; Devor, M. Chemically mediated cross-excitation in rat dorsal root ganglia. J. Neurosci. 1996, 16, 4733–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.; Devor, M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience 2000, 95, 189–195. [Google Scholar] [CrossRef]
- Kim, Y.S.; Anderson, M.; Park, K.; Zheng, Q.; Agarwal, A.; Gong, C.; Saijilafu, Y.L.; He, S.; LaVinka, P.C.; Zhou, F.; et al. Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 2016, 91, 1085–1096. [Google Scholar] [CrossRef] [Green Version]
- Cairns, B.E. Pathophysiology of TMD pain-basic mechanisms and their implications for pharmacotherapy. J. Oral. Rehabil. 2010, 37, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Ashina, S.; Bendtsen, L.; Ashina, M. Pathophysiology of tension-type headache. Curr. Pain Headache Rep. 2005, 9, 415–422. [Google Scholar] [CrossRef]
- Ashina, S.; Mitsikostas, D.D.; Lee, M.J.; Yamani, N.; Wang, S.J.; Messina, R.; Ashina, H.; Buse, D.C.; Pozo-Rosich, P.; Jensen, R.H.; et al. Tension-type headache. Nat. Rev. Dis. Prim. 2021, 7, 24. [Google Scholar] [CrossRef]
- Blumenfeld, A.; McVige, J.; Knievel, K. Post-traumatic headache: Pathophysiology and management—A review. J. Concussion 2022, 6. [Google Scholar] [CrossRef]
- Edvinsson, J.C.A.; Viganó, A.; Alekseeva, A.; Alieva, E.; Arruda, R.; De Luca, C.; D’Ettore, N.; Frattale, I.; Kurnukhina, M.; Macerola, N.; et al. The fifth cranial nerve in headaches. J. Headache Pain 2020, 21, 65. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.M.; Ashghar, M.S.; Hougaard, A.; Hansen, A.E.; Larsen, V.A.; de Koning, P.J.H.; Larsson, H.B.W.; Olesen, J.; Ahin, M. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: A cross-sectional study. Lancet Neurol. 2013, 12, 454–461. [Google Scholar] [CrossRef]
- Schou, W.S.; Ashina, S.; Amin, F.M.; Goadsby, P.J.; Ashina, M. Calcitonin gene-related peptide and pain: A systematic review. J. Headache Pain 2017, 18, 34. [Google Scholar] [CrossRef] [Green Version]
- Ailani, J.; Burch, R.C.; Robbins, M.S.; Board of Directors of the American Headache Society. The American Headache Society Consensus Statement: Update on integrating new migraine treatments into clinical practice. Headache 2021, 61, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, J.C.A.; Maddahi, A.; Christiansen, I.M.; Reducha, P.V.; Warfvinge, K.; Sheykhzade, M.; Edvinsson, L.; Haanes, K.A. Lasmiditan and 5-Hydroxytryptamine in the rat trigeminal system; expression, release and interactions with 5-HT1 receptors. J. Headache Pain 2022, 23, 26. [Google Scholar] [CrossRef]
- Clemow, D.B.; Johnson, K.W.; Hochstetler, H.M.; Ossipov, M.H.; Hake, A.M.; Blumenfeld, A.M. Lasmiditan mechanism of action—Review of a selective 5-HT1F agonist. J. Headache Pain 2020, 21, 71. [Google Scholar] [CrossRef]
- Ho, T.W.; Mannix, L.K.; Fan, X.; Assaid, C.; Furtek, C.; Jones, C.J.; Lines, C.R.; Rapoport, A.M. Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. Neurology 2008, 70, 1304–1312. [Google Scholar] [CrossRef]
- Christensen, S.L.; Petersen, S.; Kristensen, D.M.; Olesen, J.; Munro, G. Targeting CGRP via receptor antagonism and antibody neutralisation in two distinct rodent models of migraine-like pain. Cephalalgia 2019, 39, 1827–1837. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Francavilla, M.; Zanaboni, A.M.; Tassorelli, C. Antagonism of CGRP Receptor: Central and Peripheral Mechanisms and Mediators in an Animal Model of Chronic Migraine. Cells 2022, 11, 3092. [Google Scholar] [CrossRef]
- Sato, J.; Segami, N.; Kaneyama, K.; Yoshimura, H.; Fujimura, K.; Yoshitake, Y. Relationship of calcitonin gene-related peptide in synovial tissues and temporomandibular joint pain in humans. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2004, 98, 533–540. [Google Scholar] [CrossRef]
- Christensen, S.L.; Ernstsen, C.; Olesen, J.; Kristensen, D.M. No central action of CGRP antagonising drugs in the GTN mouse model of migraine. Cephalalgia 2020, 40, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Durham, P.L.; Niemann, C.; Cady, R. Repression of stimulated calcitonin gene-related peptide secretion by topiramate. Headache 2006, 46, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Durham, P.L.; Cady, R.; Cady, R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache 2004, 44, 35–42. [Google Scholar] [CrossRef]
- Shao, Y.-F.; Zhang, Y.; Zhao, P.; Yan, W.J.; Kong, X.P.; Fan, L.L.; Hou, Y.P. Botulinum Toxin Type A Therapy in Migraine: Preclin-ical and Clinical Trials. Iran. Red. Crescent. Med. J. 2013, 15, e7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuteri, D.; Tonin, P.; Nicotera, P.; Vulnera, M.; Altieri, G.C.; Tarsitano, A.; Bagetta, G.; Corasaniti, M.T. Pooled Analysis of Real-World Evidence Supports Anti-CGRP mAbs and OnabotulinumtoxinA Combined Trial in Chronic Migraine. Toxins 2022, 14, 529. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Dermitzakis, E.V.; Xiromerisiou, G.; Vikelis, M. OnabotulinumtoxinA Add-On to Monoclonal Anti-CGRP Antibodies in Treatment-Refractory Chronic Migraine. Toxins 2022, 14, 847. [Google Scholar] [CrossRef] [PubMed]
- Mechtler, L.; Saikali, N.; McVige, J.; Hughes, O.; Traut, A.; Adams, A.M. Real-World Evidence for the Safety and Efficacy of CGRP Monoclonal Antibody Therapy Added to OnabotulinumtoxinA Treatment for Migraine Prevention in Adult Patients with Chronic Migraine. Front. Neurol. 2022, 12, 788159. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, S.; Cho, S.; Kang, E.S.; Chung, C.S. Feasibility of serum CGRP measurement as a biomarker of chronic migraine: A critical reappraisal. J. Headache Pain 2018, 19, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchivileva, I.E.; Johnson, K.W.; Chai, X.; VanDam, L.R.; Lim, P.F.; Slade, G.D. Evaluation of Plasma Calcitonin Gene-Related Peptide as a Biomarker for Painful Temporomandibular Disorder and Migraine. J. Pain Res. 2023, 16, 2331–2346. [Google Scholar] [CrossRef] [PubMed]
- Karsan, N.; Goadsby, P.J. Calcitonin gene-related peptide and migraine. Curr. Opin. Neurol. 2015, 28, 250–254. [Google Scholar] [CrossRef]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP may play a causative role in mi-graine. Cephalalgia Int. J. Headache 2002, 22, 54–61. [Google Scholar] [CrossRef]
- Hansen, J.M.; Hauge, A.; Olesen, J.; Ashina, M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia 2010, 30, 1179–1186. [Google Scholar] [CrossRef]
- Cernuda-Morollón, E.; Larrosa, D.; Ramón, C.; Vega, J.; Martínez-Camblor, P.; Pascual, J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 2013, 81, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanar, R.; Dessem, D.; Moutanni, A.; Yallampalli, C.; Yallampalli, U.; Gangula, P.; Bai, G. Muscle inflammation induc-es a rapid increase in calcitonin gene-related peptide (CGRP) mRNA that temporally relates to CGRP immunoreactivity and nociceptive behavior. Neuroscience 2006, 143, 875–884. [Google Scholar] [CrossRef]
- Akerman, S.; Romero-Reyes, M. Preclinical studies investigating the neural mechanisms involved in the co-morbidity of mi-graine and temporomandibular disorders: The role of CGRP. Br. J. Pharmacol. 2020, 177, 5555–5568. [Google Scholar] [CrossRef]
- Körtési, T.; Tuka, B.; Nyári, A.; Vécsei, L.; Tajti, J. The effect of orofacial complete Freund’s adjuvant treatment on the expres-sion of migraine-related molecules. J. Headache Pain 2019, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Vause, C.V.; Durham, P.L. Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from tri-geminal ganglion glial cells. Brain Res. 2008, 1196, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afroz, S.; Arakaki, R.; Iwasa, T.; Oshima, M.; Hosoki, M.; Inoue, M.; Baba, O.; Okayama, Y.; Matsuka, Y. CGRP Induces Differential Regulation of Cytokines from Satellite Glial Cells in Trigeminal Ganglia and Orofacial Nociception. Int. J. Mol. Sci. 2019, 20, 711. [Google Scholar] [CrossRef] [Green Version]
- Just, T.B.; Schwulst, S.J. Temporomandibular disorders and traumatic brain injury: Two sides of the same coin. Adv. Oral. Maxillofac. Surg. 2021, 4, 00193. [Google Scholar] [CrossRef]
- Fiorentino, P.M.; Tallents, R.H.; Miller, J.H.; Brouxhon, S.M.; O’Banion, M.K.; Puzas, J.E.; Kyrkanides, S. Spinal interleukin-1beta in a mouse model of arthritis and joint pain. Arthr. Rheumat. 2008, 58, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Shaftel, S.S.; Miller, J.H.; Tallents, R.H.; Chang, Y.; Pinkert, C.A.; Olschowka, J.A.; Dickerson, I.M.; Puzas, J.E.; O’Banion, M.K.; et al. Intraarticular induction of interleukin-1beta expression in the adult mouse, with resultant temporomandibu-lar joint pathologic changes, dysfunction, and pain. Arthr Rheumat. 2006, 54, 1184–1197. [Google Scholar] [CrossRef] [PubMed]
- Brouxhon, S.M.; O’Banion, M.K.; Dickerson, I.M.; Kyrkanides, S. Calcitonin gene-related pep-tide: An intra-articular thera-peutic target for TMJ disorders. Clin. Exp. Dent. Res. 2022, 8, 1158–1166. [Google Scholar] [CrossRef]
- Shu, H.; Liu, S.; Tang, Y.; Schmidt, B.L.; Dolan, J.C.; Bellinger, L.L.; Kramer, P.R.; Bender, S.D.; Tao, F. A Pre-Existing Myogenic Temporomandibular Dis-order Increases Trigeminal Calcitonin Gene-Related Peptide and Enhances Nitroglycerin-Induced Hypersensitivity in Mice. Int. J. Mol. Sci. 2020, 21, 4049. [Google Scholar] [CrossRef] [PubMed]
- Damico, J.P.; Ervolino, E.; Torres, K.R.; Sabino Batagello, D.; Cruz-Rizzolo, R.J.; Aparecido Casatti, C.; Bauer, J.A. Phenotypic al-terations of neuropeptide Y and calcitonin gene-related peptide-containing neurons innervating the rat temporomandibular joint during carrageenan-induced arthritis. Eur. J. Histochem. 2012, 56, e31. [Google Scholar] [CrossRef] [Green Version]
- Suttle, A.; Wang, P.; Dias, F.C.; Zhang, Q.; Luo, Q.; Simmons, L.; Bortsov, A.; Tchivileva, I.E.; Nackley, A.G.; Chen, Y. Sensory Neuron-TRPV4 Modulates Temporomandibu-lar Disorder Pain Via CGRP in Mice. J. Pain 2022, 24, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Romero-Reyes, M.; Pardi, V.; Akerman, S. A potent and selective calcitonin gene-related peptide (CGRP) receptor antagonist, MK-8825, inhibits responses to nociceptive trigeminal activation: Role of CGRP in orofacial pain. Exp. Neurol. 2015, 271, 95–103. [Google Scholar] [CrossRef]
- Messlinger, K. The big CGRP flood—Sources, sinks and signalling sites in the trigeminovascular system. J. Headache Pain 2018, 19, 22. [Google Scholar] [CrossRef] [Green Version]
- Durham, P.L.; Masterson, C.G. Two mechanisms involved in trigeminal CGRP release: Implications for migraine treatment. Headache 2013, 53, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Korczeniewska, O.A.; Kohli, D.; Benoliel, R.; Baddireddy, S.M.; Eliav, E. Pathophysiology of Post-Traumatic Trigeminal Neuropathic Pain. Biomolecules 2022, 12, 1753. [Google Scholar] [CrossRef] [PubMed]
- Labastida-Ramírez, A.; Benemei, S.; Albanese, M.; D’Amico, A.; Grillo, G.; Grosu, O.; Ertem, D.H.; Mecklenburg, J.; Fedorova, E.P.; Řehulka, P.; et al. Persistent post-traumatic head-ache: A migrainous loop or not? The clinical evidence. J. Headache Pain 2020, 21, 55. [Google Scholar] [CrossRef]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152 (Suppl. S3), S2–S15. [Google Scholar] [CrossRef]
- Bree, D.; Levy, D. Development of CGRP-dependent pain and headache related behaviours in a rat model of concussion: Im-plications for mechanisms of post-traumatic headache. Cephalalgia 2018, 38, 246–258. [Google Scholar] [CrossRef]
- Kopruszinski, C.M.; Runes, J.M.; Swiokla, J.; Weinstein, T.J.; Schwedt, T.J.; Dodick, D.W.; Anderson, T.; Navratilova, E.; Porreca, F. CGRP monoclonal antibody prevents the loss of diffuse noxious inhibitory controls (DNIC) in a mouse model of post-traumatic headache. Cephalalgia Int. J. Headache 2021, 41, 749–759. [Google Scholar] [CrossRef]
- Navratilova, E.; Rau, J.; Oyarzo, J.; Tien, J.; Mackenzie, K.; Stratton, J.; Remeniuk, B.; Schwedt, T.; Anderson, T.; Dodick, D.; et al. CGRP-dependent and independent mechanisms of acute and persistent post-traumatic headache following mild traumatic brain injury in mice. Cephalalgia Int. J. Headache 2019, 39, 1762–1775. [Google Scholar] [CrossRef]
- Wilson, J.M.; Haliwa, I.; Lee, J.; Shook, N.J. The role of dispositional mindfulness in the fear-avoidance model of pain. PLOSOne 2023, 18, e0280740. [Google Scholar] [CrossRef] [PubMed]
- Ashina, H.; Iljazi, A.; Al-Khazali, H.M.; Eigenbrodt, A.K.; Larsen, E.L.; Andersen, A.M.; Hansen, K.J.; Bräuner, K.B.; Mørch-Jessen, T.; Chaudhry, B.; et al. Efficacy, tolerability, and safety of erenumab for the preventive treatment of persistent post-traumatic headache attributed to mild traumatic brain in-jury: An open-label study. J. Headache Pain 2020, 21, 62. [Google Scholar] [CrossRef]
- Fricton, J.; Eli, B.; Gupta, A.; Johnson, N. Preventing chronic pain after acute jaw sprain or strain. J. Am. Dent. Assoc. 2016, 147, 979–986. [Google Scholar] [CrossRef] [PubMed]
- McVige, J.; Rooney, M.; Lis, D. Anti-Calcitonin Gene-Related Peptide Monoclonal Antibodies in the Treatment of Patients With Concussion. Neurology 2022, 98, S9. [Google Scholar] [CrossRef]
- Ma, X.; Yue, Z.Q.; Gong, Z.-Q.; Zhang, H.; Duan, N.-Y.; Shi, Y.-T.; Wei, G.X.; Li, Y.F. The Effect of Diaphragmatic Breathing on Attention, Negative Affect and Stress in Healthy Adults. Front. Psychol. 2017, 8, 874. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.; Loveman, E.; Clegg, A.; Easton, S.; Berry, N. Systematic review of cognitive behavioural therapy for the management of headaches and migraines in adults. Br. J. Pain 2015, 9, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Macea, D.D.; Gajos, K.; Calil, Y.A.D.; Fregni, F. The Efficacy of Web-Based Cognitive Behavioral Interventions for Chronic Pain: A Systematic Review and Meta-Analysis. J. Pain 2010, 11, 917–929. [Google Scholar] [CrossRef]
- Turner, J.A.; Holtman, S.; Mancl, L. Mediators, moderators, and predictors of therapeutic change in cognitive-behavioral therapy for chronic pain. Pain 2007, 127, 276–286. [Google Scholar] [CrossRef]
- Fricton, J.R.; Ouyang, W.; Nixdorf, D.R.; Schiffman, E.L.; Velly, A.M.; Look, J.O. Critical appraisal of methods used in randomized controlled trials of treatments for temporomandibular disorders. J. Orofac. Pain 2010, 24, 139–151. [Google Scholar] [PubMed]
- Finnegan, E.; Daly, E.; Pearce, A.J.; Ryan, L. Nutritional interventions to support acute mTBI recovery. Front. Nutr. 2022, 9, 977728. [Google Scholar] [CrossRef]
- Fila, M.; Chojnacki, J.; Sobczuk, P.; Chojnacki, C.; Blasiak, J. Nutrition and Calcitonin Gene Related Peptide (CGRP) in Migraine. Nutrients 2023, 15, 289. [Google Scholar] [CrossRef]
- Shoeibi, A.; Olfati, N.; Soltani Sabi, M.; Salehi, M.; Mali, S.; Akbari Oryani, M. Effectiveness of coenzyme Q10 in prophylactic treatment of migraine headache: An open-label, add-on, controlled trial. Acta Neurol. Belg. 2017, 117, 103–109. [Google Scholar] [CrossRef]
- Slater, S.K.; Nelson, T.; Kabbouche, M.A.; LeCates, S.L.; Horn, P.; Segers, A.; Manning, P.; Powers, S.W.; Hershey, A.D. A randomized, double-blinded, placebo-controlled, crossover, add-on study of CoEnzyme Q10 in the prevention of pediatric and adolescent migraine. Cephalalgia 2011, 31, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Dahri, M.; Tarighat-Esfanjani, A.; Asghari-Jafarabadi, M.; Hashemilar, M. Oral coenzyme Q10 supplementation in patients with migraine: Effects on clinical features and inflammatory markers. Nutr. Neurosci. 2019, 22, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Parohan, M.; Sarraf, P.; Javanbakht, M.H.; Foroushani, A.R.; Ranji-Burachaloo, S.; Djalali, M. The synergistic effects of nano-curcumin and coenzyme Q10 supplementation in migraine prophylaxis: A randomized, placebo-controlled, double-blind trial. Nutr. Neurosci. 2021, 24, 317–326. [Google Scholar] [CrossRef]
- Hajihashemi, P.A.G.; Khorvash, F.; Reza Maracy, M.; Nourian, M. The effects of concurrent Coenzyme Q10, L-carnitine supplementation in migraine prophylaxis: A randomized, placebo-controlled, double-blind trial. Cephalalgia 2019, 39, 648–654. [Google Scholar] [CrossRef]
- Rezaie, S.; Askari, G.; Khorvash, F.; Tarrahi, M.J.; Amani, R. Effects of Curcumin Supplementation on Clinical Features and Inflammation, in Migraine Patients: A Double-Blind Controlled, Placebo Randomized Clinical Trial. Int. J. Prev. Med. 2021, 12, 161. [Google Scholar]
- Ghorbani, Z.; Rafiee, P.; Fotouhi, A.; Haghighi, S.; Magham, R.R.; Ahmadi, Z.S.; Djalali, M.; Zareei, M.; Razeghi Jahromi, S.; Shahemi, S.; et al. The effects of vitamin D supplementation on interictal serum levels of calcitonin gene-related peptide (CGRP) in episodic migraine patients: Post hoc analysis of a randomized double-blind placebo-controlled trial. J. Headache Pain 2020, 21, 22. [Google Scholar] [CrossRef] [Green Version]
- Antonopoulos, S.R.; Durham, P.L. Grape seed extract suppresses calcitonin gene-related peptide secretion and upregulates expression of GAD 65/67 and GABAB receptor in primary trigeminal ganglion cultures. IBRO Neurosci. Rep. 2022, 13, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, L.E.; Chelliboina, N.; Woodman, S.E.; Durham, P.L. Dietary supplementation with grape seed extract prevents development of trigeminal sensitization and inhibits pain signaling in a preclinical chronic temporomandibular disorder model. J. Oral. Pathol. Med. 2020, 49, 514–521. [Google Scholar] [CrossRef]
- Woodman, S.E.; Antonopoulos, S.R.; Durham, P.L. Inhibition of nociception in a preclinical episodic migraine model by dietary supplementation of grape seed extract involves activation of endocannabinoid receptors. Front. Pain Res. 2022, 3, 809352. [Google Scholar] [CrossRef]
- Sándor, P.S.; Di Clemente, L.; Coppola, G.; Saenger, U.; Fumal, A.; Magis, D.; Seidel, L.; Agosti, R.M.; Schoenen, J. Efficacy of coenzyme Q10 in migraine prophylaxis: A randomized controlled trial. Neurology 2005, 64, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, Z.; Togha, M.; Rafiee, P.; Ahmadi, Z.S.; Rasekh Magham, R.; Djalali, M.; Shahemi, S.; Martami, F.; Zareei, M.; Razeghi Jahromi, S.; et al. Vitamin D3 might improve headache characteristics and protect against inflammation in migraine: A randomized clinical trial. Neurol. Sci. 2020, 41, 1183–1192. [Google Scholar] [CrossRef]
- Azuma, Y.; Sato, I. The localization of calcitonin gene-related peptide in the human trigeminal ganglion and masseter mus-cle. Okajimas Folia Anat. Jpn. 2017, 93, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Lindquist, K.A.; Belugin, S.; Hovhannisyan, A.H.; Corey, T.M.; Salmon, A.; Akopian, A.N. Identification of Trigeminal Sensory Neuronal Types Innervating Masseter Muscle. eNeuro 2021, 8, ENEURO.0176-21.2021. [Google Scholar] [CrossRef]
- Dawson, A.; List, T.; Ernberg, M.; Svensson, P. Assessment of proprioceptive allodynia after tooth-clenching exercises. J. Orofac. Pain 2012, 26, 39–48. [Google Scholar] [PubMed]
- Satokawa, C.; Nishiyama, A.; Suzuki, K.; Uesugi, S.; Kokai, S.; Ono, T. Evaluation of tissue oxygen saturation of the masseter muscle during standardised teeth clenching. J. Oral. Rehabil. 2020, 47, 19–26. [Google Scholar] [CrossRef]
- Blumenfeld, A.M.; Boyd, J.P. Adjunctive treatment of chronic migraine using an oral dental device: Overview and results of a randomized placebo-controlled crossover study. BMC Neurol. 2022, 22, 72. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.E.; Moman, R.N.; Barman, R.A.; Kleppel, D.J.; Eberhart, N.D.; Gerberi, D.J.; Murad, M.H.; Hooten, W.M. Effects of Slow Deep Breathing on Acute Clinical Pain in Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Evid-Based Integr. Med. 2022, 27, 2515690X221078006. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Huang, X.-Y.; Chien, C.-H.; Cheng, J.-F. The Effectiveness of Diaphragmatic Breathing Relaxation Training for Reducing Anxiety. Perspect. Psychiatr. Care. 2017, 53, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Sink, K.S.; Walker, D.L.; Yang, Y.; Davis, M. Calcitonin Gene-Related Peptide in the Bed Nucleus of the Stria Terminalis Produces an Anxiety-Like Pattern of Behavior and Increases Neural Activation in Anxiety-Related Structures. J. Neurosci. 2011, 31, 1802–1810. [Google Scholar] [CrossRef] [Green Version]
- Li, X.F.; Bowe, J.E.; Mitchell, J.C.; Brain, S.D.; Lightman, S.L.; O’Byrne, K.T. Stress-induced suppression of the gonadotro-pin-releasing hormone pulse generator in the female rat: A novel neural action for calcitonin gene-related peptide. Endocrin 2004, 145, 1556–1563. [Google Scholar] [CrossRef] [Green Version]
- Bowen, A.J.; Huang, Y.W.; Chen, J.Y.; Pauli, J.L.; Campos, C.A.; Palmiter, R.D. Topographic representation of current and future threats in the mouse nociceptive amygdala. Nat. Commun. 2023, 14, 196. [Google Scholar] [CrossRef]

{kind=link}
| Preclinical Studies | ||
|---|---|---|
| Author | Model | Findings |
| Cady et al. [15] | TMJ capsule | Injection of CGRP in the TMJ capsule stimulated the expression of proteins associated with peripheral and central sensitization in neuronal and glial cells in animal models. |
| Fiorentino et al. [64] | Osteoarthritis in a mouse model | CGRP-induced neuroinflammation contributed to histopathological modifications of the articular tissues (i.e., cartilage), leading to osteoarthritis in a mouse model. |
| Lai et al. [65] | TMJ of mouse model | Inhibition of CGRP-mediated neuroinflammation curbed the progression of TMJ damage. |
| Akerman et al. [59] | Rat model of myofascial TMD-like inflammation | Models of TMD-like inflammation resulted in neuronal activation and sensitization of dural trigeminal neurons, similar to migraine-like manifestation. Pre-administration of CGRP receptor antagonist effectively prevented these neuronal responses. |
| Brouxhon et al. [66] | TMJ of mouse model | The overexpression of CGRP in mouse models of TMJ led to the manifestation of joint anomalies and articular pathology; conversely, in a scenario of joint inflammation, the overexpression of CGRP inhibitory peptide partially led to improvement of joint pathology. |
| Shu et al. [67] | Myogenic TMD mice model | The presence of pre-existing myogenic TMD lesions caused increased central CGRP release and enhanced migraine hypersensitivity in animal models. |
| Damico et al. [68] | Acute and chronic arthritis model | Both acute and chronic arthritis were associated with significant increases in CRGP expression in the trigeminal ganglion in animal models. |
| Suttle et al. [69] | Mouse model | In naïve mouse models, the local injection of CGRP in masseters and/or TMD induced acute pain. Conversely, blockage of CGRP receptor decreased TMD pain. |
| Romero-Reyes et al. [70] | Mouse model of acute masseter pain | Selective CGRP receptor antagonist, MK-8825, was found to significantly reduce spontaneous orofacial pain behaviors in a mouse model of acute masseter pain injected with CFA. This study also supported the role of CGRP as an important neurotransmitter involved in TMD pain, although not through an inflammatory mechanism. |
| Clinical studies | ||
| Sato et al. [44] | TMJ pain vs. healthy control | Human subjects exhibited a significantly higher level of CGRP in deranged TMJ joints vs. healthy controls, with CGRP levels that are positively correlated with pain intensity scores. |
| Intervention | Scientific Basis | Description |
|---|---|---|
| Self-management training | Systematic reviews of behavioral therapies | Nutritional and dietary intervention Preventive medicine counseling Habit-reversal Mindfulness-based stress reduction Meditation and relaxation |
| Intra-oral splints | Systematic reviews of intra-oral splints | Full coverage stabilization at night Repositioning splints at night Immediate quick splints short-term Anterior bite plane short-term |
| Medications | Systematic reviews of medications | Migraine medication NSAIDs Acetaminophen Tricyclic medications Muscle relaxants |
| Physical therapies | Systematic review evidence of therapeutic exercises | Therapeutic exercises Mobilization |
| Nutritional Supplement | Scientific Rigor | N Participants | Findings |
|---|---|---|---|
| Coenzyme Q10 | Double-blind placebo-controlled RCT [92] | 45 female adults with migraine | Significant reduction in frequency (p = 0.018), headache intensity (p = 0.001) and duration (p = 0.012) compared to controls |
| Open-label match-controlled trial [90] | 80 adults with migraine | Significant reduction in frequency of monthly attacks (p < 0.001) and headache severity (p < 0.001) compared to controls | |
| Crossover double-blind placebo-controlled RCT [91] | 120 children and adolescents with migraine | Greater improvement in migraine frequency in the initial 1–4 weeks | |
| Double-blind RCT [100] | 42 adults with migraine | Significant decrease in attack frequency, headache-days and responder rate (47.6% vs. 14.4% in controls) | |
| Nano-curcumin and coenzyme Q10 | Double-blind placebo-controlled RCT [93] | 100 adults with migraine | Significant reduction in headache frequency, severity and duration in participants treated with nano-curcumin and coenzyme Q10 (p < 0.001) |
| L-carnitine and coenzyme Q10 | Double-blind placebo-controlled RCT [94] | 56 adults with migraine | Significant reduction in headache intensity (p < 0.001), duration (p < 0.001), frequency (p < 0.001) and headache diary results (p < 0.001) |
| L-carnitine, magnesium and magnesium-L-carnitine | Single-blind RCT | 133 adults with migraine | Magnesium supplementation achieved significantly higher reduction in headache frequency compared to the other groups (p = 0.008); significant reduction in migraine symptoms in all study groups with no difference among them |
| Curcumin | Double-blind placebo-controlled RCT [95] | 44 female adults with migraine | Significant reduction in headache intensity (p = 0.001) and duration (p = 0.007); no significant reduction in headache frequency (p = 0.052) |
| Vitamin D | Double-blind placebo-controlled RCT [96,101] | 80 adults with migraine | Significant reduction in migraine disability (p = 0.016), headache duration, intensity and frequency (p < 0.05) compared to controls |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sangalli, L.; Eli, B.; Mehrotra, S.; Sabagh, S.; Fricton, J. Calcitonin Gene-Related Peptide-Mediated Trigeminal Ganglionitis: The Biomolecular Link between Temporomandibular Disorders and Chronic Headaches. Int. J. Mol. Sci. 2023, 24, 12200. https://doi.org/10.3390/ijms241512200
Sangalli L, Eli B, Mehrotra S, Sabagh S, Fricton J. Calcitonin Gene-Related Peptide-Mediated Trigeminal Ganglionitis: The Biomolecular Link between Temporomandibular Disorders and Chronic Headaches. International Journal of Molecular Sciences. 2023; 24(15):12200. https://doi.org/10.3390/ijms241512200
Chicago/Turabian StyleSangalli, Linda, Bradley Eli, Sachi Mehrotra, Suzan Sabagh, and James Fricton. 2023. "Calcitonin Gene-Related Peptide-Mediated Trigeminal Ganglionitis: The Biomolecular Link between Temporomandibular Disorders and Chronic Headaches" International Journal of Molecular Sciences 24, no. 15: 12200. https://doi.org/10.3390/ijms241512200