

Dissecting the Structural Dynamics of Authentic Cholesteryl Ester Transfer Protein for the Discovery of Potential Lead Compounds: A Theoretical Study


Abstract
:1. Introduction
2. Results and Discussion
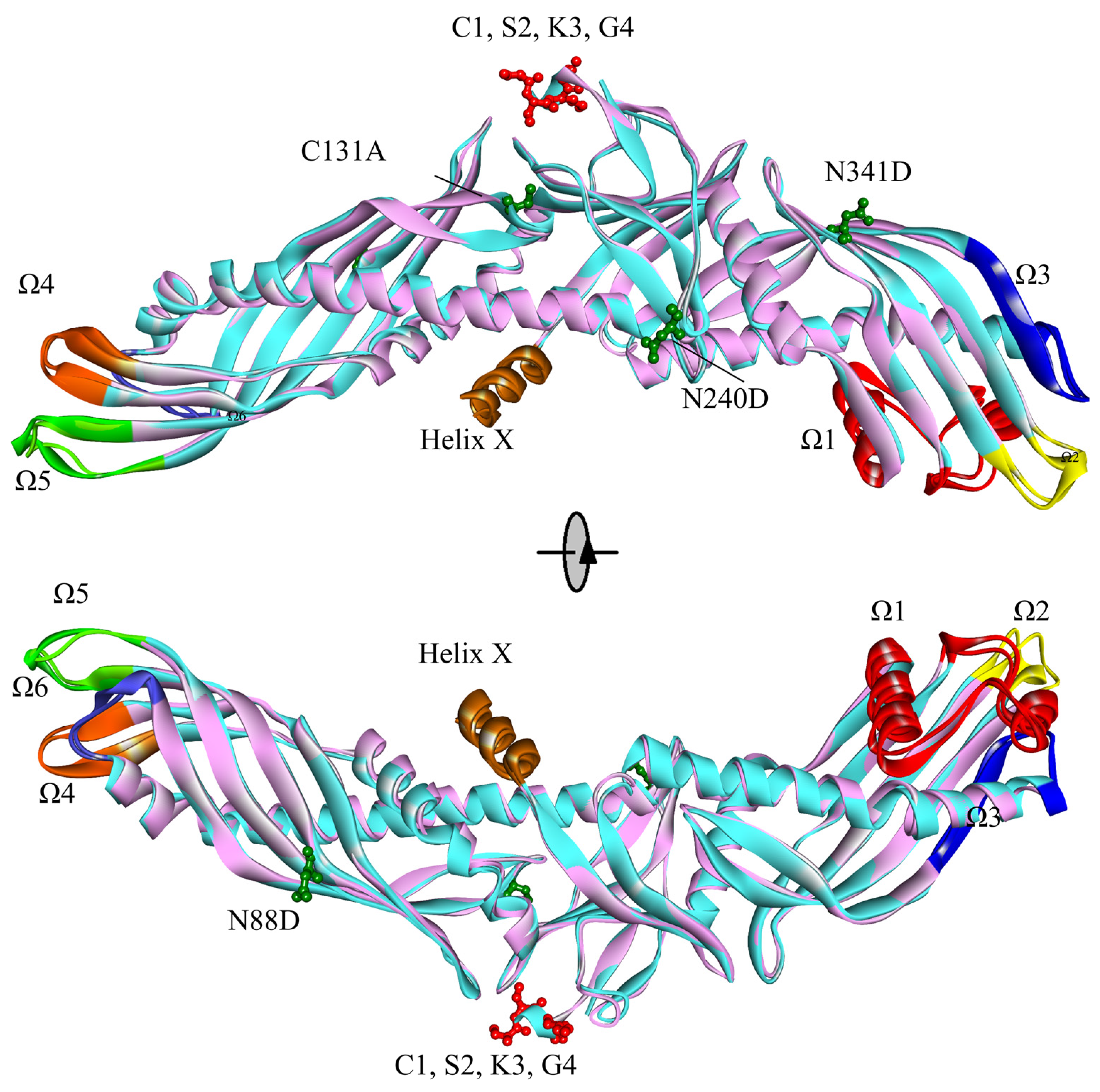
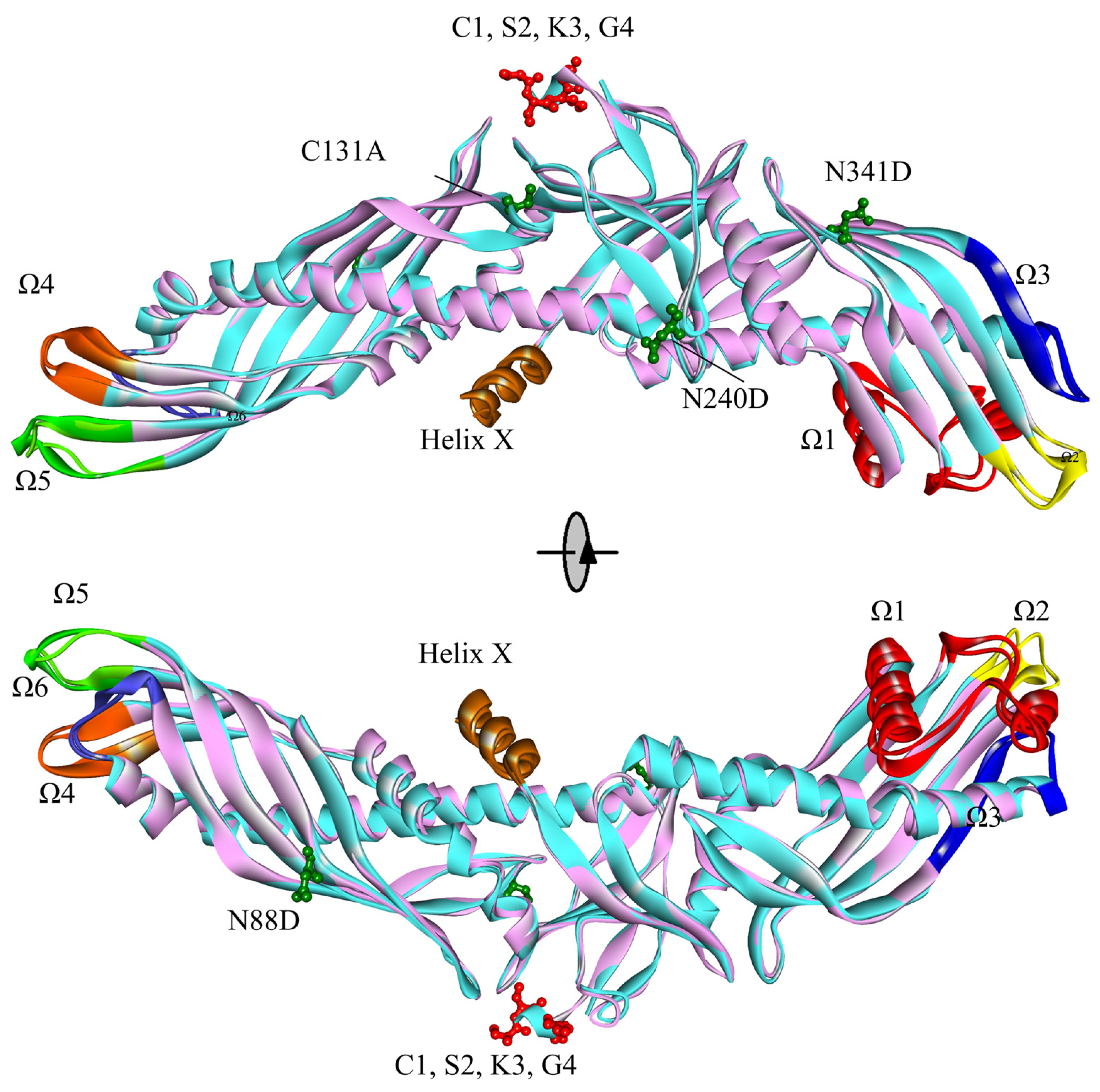
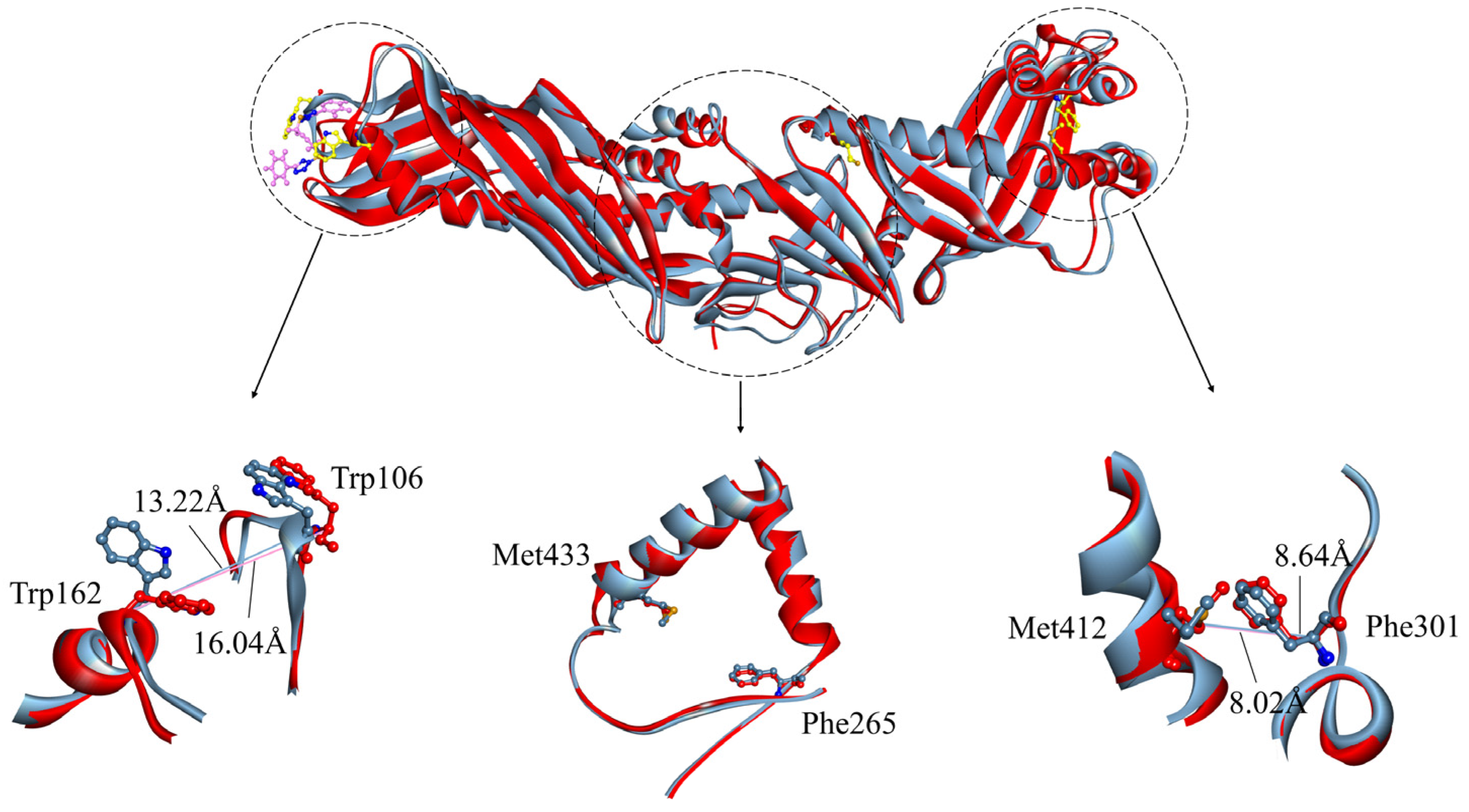
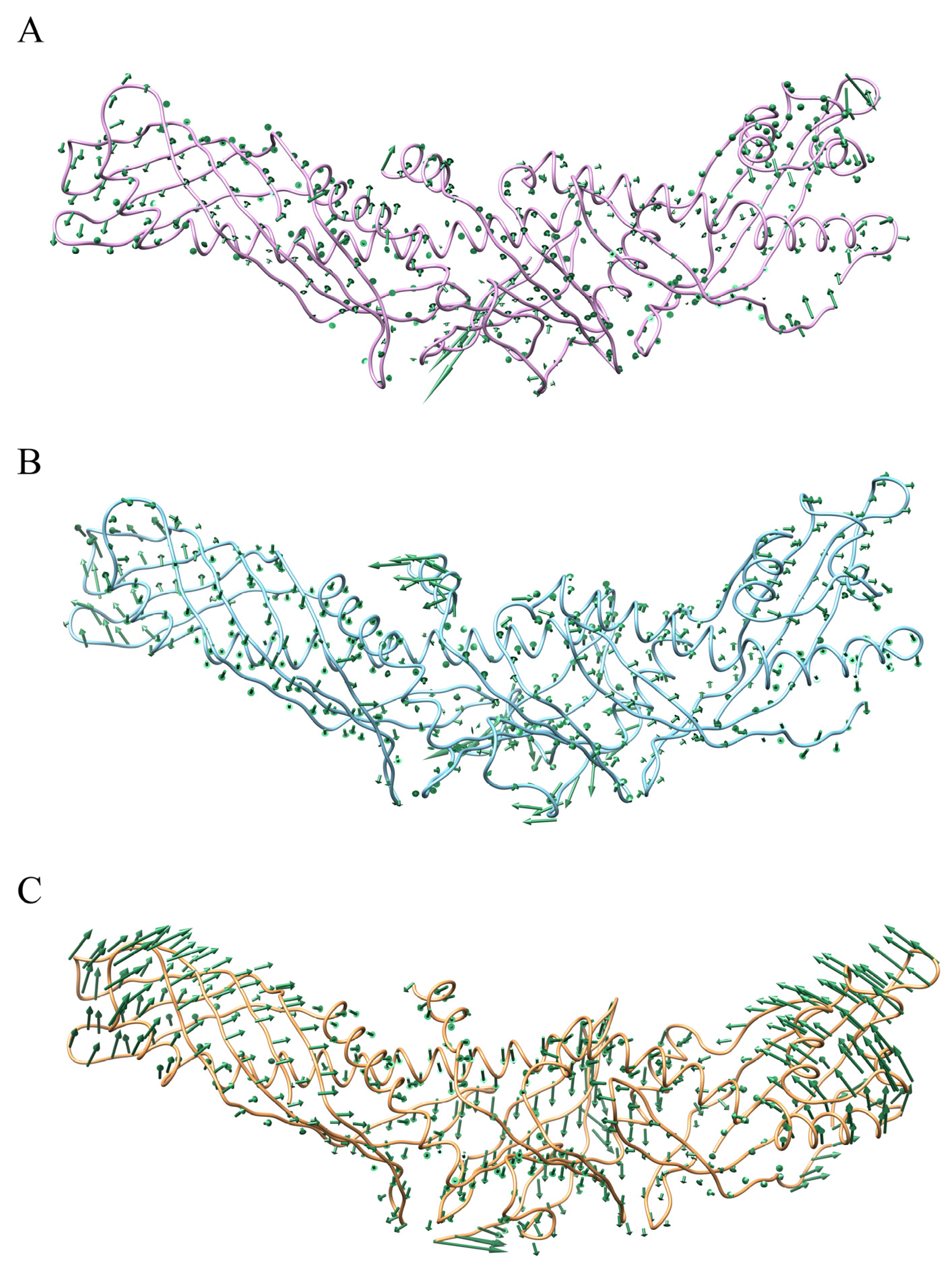
2.1. Contrast in Structural Stability and Flexibility
2.2. Contrast in the N-Terminal End, C-Terminal End, and “Neck” of the Hydrophobic Tunnel
2.3. Virtual Screening for CETP
2.4. Structural Dynamics Analysis upon Ligand Binding
3. Materials and Methods
3.1. System Preparation
3.2. MD Simulations
3.3. Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rader, D.J.; Tall, A.R. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat. Med. 2012, 18, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Jiang, X.C.; Sakai, N.; Yamashita, S.; Hirano, K.; Bujo, H.; Yamazaki, H.; Kusunoki, J.; Miura, T.; Kussie, P.; et al. A missense mutation in the cholesteryl ester transfer protein gene with possible dominant effects on plasma high density lipoproteins. J. Clin. Investig. 1993, 92, 2060–2064. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Chiba, H.; Tsuchihashi, K.; Tsuji, M.; Kumagai, M.; Matsuno, K.; Kobayashi, K. Cholesteryl ester transfer protein gene: Two common mutations and their effect on plasma high-density lipoprotein cholesterol content. J. Clin. Endocrinol. Metab. 1994, 79, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Charles, M.A.; Kane, J.P. New molecular insights into CETP structure and function: A review. J. Lipid Res. 2012, 53, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Rader, D.J.; Degoma, E.M. Future of Cholesteryl Ester Transfer Protein Inhibitors. Annu. Rev. Med. 2014, 65, 385–403. [Google Scholar] [CrossRef]
- Kingwell, B.A.; Chapman, M.J.; Kontush, A.; Miller, N.E. HDL-targeted therapies: Progress, failures and future. Nat. Rev. Drug Discov. 2014, 13, 445–464. [Google Scholar] [CrossRef]
- Yamashita, S.; Ruscica, M.; Macchi, C.; Corsini, A.; Matsuzawa, Y.; Sirtori, C.R. Cholesteryl ester transfer protein: An enigmatic pharmacology–Antagonists and agonists. Atherosclerosis 2018, 278, 286–298. [Google Scholar] [CrossRef]
- Koivuniemi, A.; Vuorela, T.; Kovanen, P.T.; Vattulainen, I.; Hyvönen, M.T. Lipid Exchange Mechanism of the Cholesteryl Ester Transfer Protein Clarified by Atomistic and Coarse-grained Simulations. PLoS Comput. Biol. 2012, 8, e1002299. [Google Scholar] [CrossRef] [Green Version]
- Karilainen, T.; Timr, S.; Vattulainen, I.; Jungwirth, P. Oxidation of Cholesterol Does Not Alter Significantly Its Uptake into High-Density Lipoprotein Particles. J. Phys. Chem. B 2015, 119, 4594–4600. [Google Scholar] [CrossRef]
- Qiu, X.; Mistry, A.; Ammirati, M.J.; Chrunyk, B.A.; Clark, R.W.; Cong, Y.; Culp, J.S.; Danley, D.E.; Freeman, T.B.; Geoghegan, K.F.; et al. Faculty Opinions recommendation of Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat. Struct. Mol. Biol. 2007, 14, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, F.; Zhang, S.; Lei, D.; Charles, M.A.; Cavigiolio, G.; Oda, M.; Krauss, R.M.; Weisgraber, K.H.; Rye, K.-A.; et al. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat. Chem. Biol. 2012, 8, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, D.; Zhang, X.; Jiang, S.; Cai, Z.; Rames, M.J.; Zhang, L.; Ren, G.; Zhang, S. Structural features of cholesteryl ester transfer protein: A molecular dynamics simulation study. Proteins Struct. Funct. Bioinform. 2013, 81, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Charles, R.; Tong, H.; Zhang, L.; Patel, M.; Wang, F.; Rames, M.J.; Ren, A.; Rye, K.A.; Qiu, X.; et al. HDL surface lipids mediate CETP binding as revealed by electron microscopy and mo-lecular dynamics simulation. Sci. Rep. 2015, 5, 8741. [Google Scholar] [CrossRef] [Green Version]
- Cilpa-Karhu, G.; Jauhiainen, M.; Riekkola, M.-L. Atomistic MD simulation reveals the mechanism by which CETP penetrates into HDL enabling lipid transfer from HDL to CETP. J. Lipid Res. 2015, 56, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barter, P.J.; Jones, M.E. Kinetic studies of the transfer of esterified cholesterol between human plasma low and high density lipoproteins. J. Lipid Res. 1980, 21, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Tall, A. Plasma-Lipid Transfer Proteins. Annu. Rev. Biochem. 1995, 64, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters, and cholesterol efflux: Implica-tions for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.L.; Inazu, A.; Hesler, C.B.; Agellon, L.B.; Mann, C.; Whitlock, M.E.; Marcel, Y.L.; Milne, R.W.; Koizumi, J.; Mabuchi, H.; et al. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature 1989, 342, 448–451. [Google Scholar] [CrossRef]
- Xue, H.; Zhang, M.; Liu, J.; Wang, J.; Ren, G. Structure-based mechanism and inhibition of cholesteryl ester transfer protein. Curr. Atheroscler. Rep. 2023, 25, 155–166. [Google Scholar] [CrossRef]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein-A novel target for raising HDL and inhibiting atherosclerosis. Arterioscl. Throm. Vas. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Jamalan, M.; Zeinali, M.; Ghaffari, M.A. A molecular dynamics investigation on the inhibition mechanism of cholesteryl ester transfer protein by Anacetrapib. Med. Chem. Res. 2016, 25, 62–69. [Google Scholar] [CrossRef]
- Chirasani, V.R.; Sankar, R.; Senapati, S. Mechanism of Inhibition of Cholesteryl Ester Transfer Protein by Small Molecule Inhibitors. J. Phys. Chem. B 2016, 120, 8254–8263. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.W.; Cao, Y.; Hao, D.X.; Yuan, X.H.; Zhang, L.; Zhang, S.L. Binding profiles of cholesterol ester transfer protein with current inhibitors: A look at mechanism and drawback. J. Biomol. Struct. Dyn. 2018, 36, 2567–2580. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Janson, C.A. Structure of apo acyl carrier protein and a proposal to engineer protein crystallization through metal ions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1545–1554. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, X.; Deng, L.; Rassart, E.; Milne, R.W.; Tall, A.R. Point mutagenesis of carboxyl-terminal amino acids of cholesteryl ester transfer protein. Opposite faces of an amphipathic helix important for cholesteryl ester transfer or for binding neutralizing antibody. J. Biol. Chem. 1993, 268, 1955–1959. [Google Scholar] [CrossRef]
- Wang, S.; Kussie, P.; Deng, L.; Tall, A. Defective binding of neutral lipids by a carboxyl-terminal deletion mutant of cho-lesteryl ester transfer protein. Evidence for a carboxyl-terminal cholesteryl ester binding site essential for neutral lipid transfer activity. J. Biol. Chem. 1995, 270, 612–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dergunov, A.D. Prediction of the influences of missense mutations on cholesteryl ester transfer protein structure. Arch. Biochem. Biophys. 2014, 564, 67–73. [Google Scholar] [CrossRef]
- Liu, S.P.; Mistry, A.; Reynolds, J.M.; Lloyd, D.B.; Griffor, M.C.; Perry, D.A.; Ruggeri, R.B.; Clark, R.W.; Qiu, X.Y. Crystal Structures of Cholesteryl Ester Transfer Protein in Complex with Inhibitors. J. Biol. Chem. 2012, 287, 37321–37329. [Google Scholar] [CrossRef] [Green Version]
- Hao, D.; Wang, H.; Zang, Y.; Zhang, L.; Yang, Z.; Zhang, S. Mechanism of Glycans Modulating Cholesteryl Ester Transfer Protein: Unveiled by Molecular Dynamics Simulation. J. Chem. Inf. Model. 2021, 62, 5246–5257. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Bi-ology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Grossfield, A.; Zuckerman, D.M. Quantifying Uncertainty and Sampling Quality in Biomolecular Simulations. Annu. Rep. Comput. Chem. 2009, 5, 23–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Äijänen, T.; Koivuniemi, A.; Javanainen, M.; Rissanen, S.; Rog, T.; Vattulainen, I. How Anacetrapib Inhibits the Activity of the Cholesteryl Ester Transfer Protein? Perspective through Atomistic Simulations. PLoS Comput. Biol. 2014, 10, e1003987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.; Bennett, W.S., Jr. Functional significance of flexibility in proteins. Biopolymers 1983, 22, 261–279. [Google Scholar] [CrossRef]
- Gerstein, M.; Lesk, A.M.; Chothia, C. Structural Mechanisms for Domain Movements in Proteins. Biochemistry 1994, 33, 6739–6749. [Google Scholar] [CrossRef]
- Zheng, K.-Q.; Zhang, S.-Z.; Zhang, L.; Huang, D.-J.; Liao, L.-C.; Hou, Y.-P. A Novel Missense Mutation (L296Q) in Cholesteryl Ester Transfer Protein Gene Related to Coronary Heart Disease. Acta Biochim. Biophys. Sin. 2004, 36, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Lei, D.S.; Tong, H.M.; Zhang, L.; Zhang, X.; Zhang, S.L.; Ren, G. Structure and Function of Cholesteryl Ester Transfer Protein in Transferring Cholesteryl Ester. Prog. Chem. 2014, 26, 879–888. [Google Scholar]
- Wang, S.; Deng, L.P.; Milne, R.W.; Tall, A.R. Identification of a sequence within the C-terminal 26 amino acids of cholesteryl ester transfer protein responsible for binding a neutralizing monoclonal antibody and necessary for neutral lipid transfer activity. J. Biol. Chem. 1992, 267, 17487–17490. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [Green Version]
- Chirasani, V.R.; Revanasiddappa, P.D.; Senapati, S. Structural Plasticity of Cholesteryl Ester Transfer Protein Assists the Lipid Transfer Activity. J. Biol. Chem. 2016, 291, 19462–19473. [Google Scholar] [CrossRef] [Green Version]
- Accelrys Discovery Studio 3.1. Available online: http://accelrys.com (accessed on 8 March 2023).
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Dong, B.-L.; Liao, Q.-H.; Wei, J. Docking and molecular dynamics study on the inhibitory activity of N, N-disubstituted-trifluoro-3-amino-2-propanols-based inhibitors of cholesteryl ester transfer protein. J. Mol. Model. 2010, 17, 1727–1734. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. Charmm-a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Hao, D.X.; Yang, Z.W.; Gao, T.; Tian, Z.Q.; Zhang, L.; Zhang, S.L. Role of glycans in cholesteryl ester transfer protein revealed by molecular dynamics simulation. Proteins Struct. Funct. Bioinform. 2018, 86, 882–891. [Google Scholar] [CrossRef]
- Xia, J.; Yang, L.; Dong, L.; Niu, M.; Zhang, S.; Yang, Z.; Wumaier, G.; Li, Y.; Wei, X.; Gong, Y.; et al. Cefminox, a Dual Agonist of Prostacyclin Receptor and Peroxisome Proliferator-Activated Receptor-Gamma Identified by Virtual Screening, Has Therapeutic Efficacy against Hypoxia-Induced Pulmonary Hypertension in Rats. Front. Pharmacol. 2018, 9, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhao, Y.; Hao, D.; Wang, H.; Li, S.; Jia, L.; Yuan, X.; Zhang, L.; Meng, L.; Zhang, S. Computational identification of potential chemoprophylactic agents according to dynamic behavior of peroxisome proliferator-activated receptor gamma. Rsc. Adv. 2021, 11, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhu, X.; Zhao, Y.Z.; Zang, Y.J.; Zhang, J.W.; Kang, Y.; Yang, Z.W.; Lin, P.; Zhang, L.; Zhang, S.L. Markov State Models Underlying the N-Terminal Premodel of TOPK/PBK. J. Phys. Chem. B 2022, 126, 10662–10671. [Google Scholar] [CrossRef]
- Yang, Z.W.; Zhao, Y.-Z.; Zang, Y.-J.; Wang, H.; Zhu, X.; Meng, L.-J.; Yuan, X.-H.; Zhang, L.; Zhang, S.-L. Rapid Structure-Based Screening Informs Potential Agents for Coronavirus Disease (COVID-19) Outbreak*. Chin. Phys. Lett. 2020, 37, 058701. [Google Scholar] [CrossRef]
- Yang, Z.W.; Zang, Y.J.; Wang, H.; Kang, Y.; Zhang, J.W.; Li, X.H.; Zhang, L.; Zhang, S.L. Recognition between CD147 and cyclophilin A deciphered by accelerated molecular dynamics simulations. Phys. Chem. Chem. Phys. 2022, 24, 18905–18914. [Google Scholar] [CrossRef] [PubMed]
- Fataftah, H.; Karain, W. Detecting protein atom correlations using correlation of probability of recurrence. Proteins: Struct. Funct. Bioinform. 2014, 82, 2180–2189. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. Software News and Updates MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Lill, M.A.; Thompson, J.J. Solvent Interaction Energy Calculations on Molecular Dynamics Trajectories: Increasing the Efficiency Using Systematic Frame Selection. J. Chem. Inf. Model. 2011, 51, 2680–2689. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CETPMutant | CETPAuthentic | |||||
|---|---|---|---|---|---|---|
| Region | Hydrophobic | Hydrophilic | Total | Hydrophobic | Hydrophilic | Total |
| N-end 2 | −0.05 | 0.02 | −0.04 | −0.05 | 0.00 | −0.02 |
| C-end 3 | −0.02 | 0.08 | 0.05 | 0.07 | 0.08 | 0.08 |
| Ω1 | −0.13 | 0.09 | −0.05 | 0.04 | 0.04 | 0.04 |
| Ω2 | 0.15 | 0.03 | 0.18 | 0.19 | 0.18 | 0.19 |
| Ω3 | 0.05 | 0.08 | 0.12 | 0.05 | 0.10 | 0.09 |
| Ω4 | 0.08 | 0.05 | 0.13 | 0.04 | 0.01 | 0.02 |
| Ω5 | −0.08 | −0.07 | −0.15 | −0.08 | −0.06 | −0.07 |
| Ω6 | −0.15 | 0.07 | −0.08 | −0.10 | 0.05 | 0.00 |
| Helix X | 0.02 | 0.03 | 0.06 | 0.10 | 0.01 | 0.03 |
| ZINC ID | ΔEele 2 | ΔEvdw 3 | ΔGsur 4 | ΔGGB 5 | ΔGbind |
|---|---|---|---|---|---|
| ZINC000002010603 | −2.2 ± 1.5 | −14.2 ± 0.9 | −8.9 ± 0.5 | 1.7 ± 0.1 | −23.6 ± 0.2 |
| ZINC000006248133 | −5.5 ± 2.0 | −12.6 ± 1.0 | −8.4 ± 0.7 | 1.1 ± 1.5 | −25.4 ± 2.1 |
| ZINC000005871812 | −5.6 ± 1.8 | −13.1 ± 1.1 | −8.7 ± 0.7 | 2.0 ± 1.3 | −25.4 ± 2.1 |
| ZINC000002261174 | −5.8 ± 1.4 | −19.5 ± 0.9 | −12.3 ± 0.6 | 2.5 ± 1.0 | −35.0 ± 1.7 |
| ZINC000003526223 | 0.0 ± 0.5 | −0.1 ± 0.9 | −0.1 ± 0.7 | −0.1 ± 0.4 | −0.2 ± 0.2 |
| ZINC000005871644 | −2.6 ± 1.4 | −14.9 ± 1.2 | −9.7 ± 0.8 | 0.9 ± 1.1 | −26.3 ± 2.3 |
| ZINC000007067674 | −2.2 ± 1.4 | −11.3 ± 1.5 | −7.0 ± 0.1 | 0.7 ± 1.1 | −19.7 ± 2.8 |
| ZINC000006242926 | −2.5 ± 1.1 | −19.5 ± 0.8 | −11.2 ± 0.5 | 0.6 ± 0.9 | −32.6 ± 1.5 |
| ZINC ID | ΔEele 2 | ΔEvdw 3 | ΔGsur 4 | ΔGGB 5 | ΔGbind |
|---|---|---|---|---|---|
| ZINC000002010603 | −5.8 ± 3.1 | −17.5 ± 1.1 | −10.1 ± 0.7 | 0.4 ± 2.7 | −32.9 ± 2.3 |
| ZINC000006248133 | −9.1 ± 2.3 | −19.6 ± 0.8 | −13.0 ± 0.6 | 3.2 ± 2.0 | −38.8 ± 1.6 |
| ZINC000005871812 | −2.8 ± 1.6 | −26.6 ± 0.8 | −15.5 ± 0.4 | −0.4 ± 1.3 | −45.3 ± 1.3 |
| ZINC000002261174 | −2.7 ± 1.3 | −19.0 ± 0.9 | −11.5 ± 0.6 | −0.3 ± 1.1 | −33.5 ± 1.8 |
| ZINC000003526223 | −7.0 ± 1.7 | −28.5 ± 0.8 | −16.5 ± 0.5 | 5.5 ± 1.6 | −46.5 ± 1.3 |
| ZINC000005871644 | −5.1 ± 1.6 | −23.1 ± 0.8 | −14.3 ± 0.5 | 3.1 ± 1.4 | −39.4 ± 1.4 |
| ZINC000007067674 | −2.3 ± 1.2 | −26.1 ± 0.6 | −15.6 ± 0.3 | 0.6 ± 1.1 | −43.4 ± 1.0 |
| ZINC000006242926 | −1.4 ± 1.2 | −25.9 ± 0.9 | −15.0 ± 0.5 | 0.1 ± 1.1 | −42.2 ± 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Hao, D.; Zhao, Y.; Zhang, S.; Zhang, L.; Yang, Z. Dissecting the Structural Dynamics of Authentic Cholesteryl Ester Transfer Protein for the Discovery of Potential Lead Compounds: A Theoretical Study. Int. J. Mol. Sci. 2023, 24, 12252. https://doi.org/10.3390/ijms241512252
Zhao Y, Hao D, Zhao Y, Zhang S, Zhang L, Yang Z. Dissecting the Structural Dynamics of Authentic Cholesteryl Ester Transfer Protein for the Discovery of Potential Lead Compounds: A Theoretical Study. International Journal of Molecular Sciences. 2023; 24(15):12252. https://doi.org/10.3390/ijms241512252
Chicago/Turabian StyleZhao, Yizhen, Dongxiao Hao, Yifan Zhao, Shengli Zhang, Lei Zhang, and Zhiwei Yang. 2023. "Dissecting the Structural Dynamics of Authentic Cholesteryl Ester Transfer Protein for the Discovery of Potential Lead Compounds: A Theoretical Study" International Journal of Molecular Sciences 24, no. 15: 12252. https://doi.org/10.3390/ijms241512252
APA StyleZhao, Y., Hao, D., Zhao, Y., Zhang, S., Zhang, L., & Yang, Z. (2023). Dissecting the Structural Dynamics of Authentic Cholesteryl Ester Transfer Protein for the Discovery of Potential Lead Compounds: A Theoretical Study. International Journal of Molecular Sciences, 24(15), 12252. https://doi.org/10.3390/ijms241512252