
Mouse Models of Cardiomyopathies Caused by Mutations in Troponin C

Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of cTnC in the Regulation of Cardiac Muscle Contraction
3. Why Focus on cTnC
4. Ca2+ Exchange Kinetics of cTnC
5. What Can Go Wrong with cTnC Function?
6. How Might cTnC Mutations Lead to Disease?
7. Known HCM-, RCM-, DCM, and LVNC-Associated Mutations in Human cTnC
8. Mutations in cTnC Linked to HCM
9. Mutations in cTnC Linked to RCM
10. Mutations in cTnC Linked to DCM
11. Mutations in cTnC Linked to LVNC
12. Summary of the Data on Human Patients
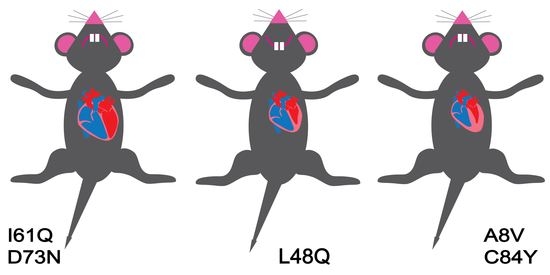
13. Genetically Engineered Mouse Models
14. Summary of the Data in Genetically Engineered Mice
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciarambino, T.; Menna, G.; Sansone, G.; Giordano, M. Cardiomyopathies: An Overview. Int. J. Mol. Sci. 2021, 22, 7722. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Cardiomyopathies: An Overview. Circ. Res. 2017, 121, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.J.; Tardiff, J.C. Complexity in genetic cardiomyopathies and new approaches for mechanism-based precision medicine. J. Gen. Physiol. 2021, 153, e202012662. [Google Scholar] [CrossRef] [PubMed]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Silva Enciso, J.; Tardiff, J.C.; Adler, E.D. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef]
- Deranek, A.E.; Klass, M.M.; Tardiff, J.C. Moving beyond simple answers to complex disorders in sarcomeric cardiomyopathies: The role of integrated systems. Pflugers Arch. 2019, 471, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Massera, D.; Sherrid, M.V.; Maron, M.S.; Rowin, E.J.; Maron, B.J. How common is hypertrophic cardiomyopathy... really?: Disease prevalence revisited 27 years after CARDIA. Int. J. Cardiol. 2023, 382, 64–67. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef]
- Barefield, D.Y.; Alvarez-Arce, A.; Araujo, K.N. Mechanisms of Sarcomere Protein Mutation-Induced Cardiomyopathies. Curr. Cardiol. Rep. 2023, 25, 473–484. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Rapezzi, C.; Aimo, A.; Barison, A.; Emdin, M.; Porcari, A.; Linhart, A.; Keren, A.; Merlo, M.; Sinagra, G. Restrictive cardiomyopathy: Definition and diagnosis. Eur. Heart J. 2022, 43, 4679–4693. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L. Left Ventricular Noncompaction Cardiomyopathy: New Clues in a Not So New Disease? J. Am. Heart Assoc. 2021, 10, e018815. [Google Scholar] [CrossRef] [PubMed]
- Mirza, H.; Mohan, G.; Khan, W.; Alkhatib, A.; Kaur, I.; Asif, M.; Shah, A.; Mughal, M.S. A Review of Left Ventricular Non-compaction Cardiomyopathy (LVNC). J. Community Hosp. Intern. Med. Perspect. 2022, 12, 51–63. [Google Scholar] [CrossRef]
- Litt, M.J.; Ali, A.; Reza, N. Familial Hypertrophic Cardiomyopathy: Diagnosis and Management. Vasc. Health Risk Manag. 2023, 19, 211–221. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef]
- Helms, A.S.; Thompson, A.D.; Day, S.M. Translation of New and Emerging Therapies for Genetic Cardiomyopathies. JACC Basic. Transl. Sci. 2022, 7, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Cimiotti, D.; Budde, H.; Hassoun, R.; Jaquet, K. Genetic Restrictive Cardiomyopathy: Causes and Consequences-An Integrative Approach. Int. J. Mol. Sci. 2021, 22, 558. [Google Scholar] [CrossRef]
- Towbin, J.A.; Jefferies, J.L. Cardiomyopathies Due to Left Ventricular Noncompaction, Mitochondrial and Storage Diseases, and Inborn Errors of Metabolism. Circ. Res. 2017, 121, 838–854. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; McKenna, W.J. Left ventricular noncompaction and cardiomyopathy: Cause, contributor, or epiphenomenon? Curr. Opin. Cardiol. 2008, 23, 171–175. [Google Scholar] [CrossRef]
- Li, M.X.; Hwang, P.M. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.P.; Tikunova, S.B. Ca(2+) exchange with troponin C and cardiac muscle dynamics. Cardiovasc. Res. 2008, 77, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.A.; Pinto, J.R.; Moraes, A.H.; Iqbal, A.; de Magalhaes, M.T.; Monteiro, J.; Pedrote, M.M.; Sorenson, M.M.; Silva, J.L.; de Oliveira, G.A. Allosteric Transmission along a Loosely Structured Backbone Allows a Cardiac Troponin C Mutant to Function with Only One Ca2+ Ion. J. Biol. Chem. 2017, 292, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.; Xu, Y.; Kirberger, M.; Yang, J.J. Structural Aspects and Prediction of Calmodulin-Binding Proteins. Int. J. Mol. Sci. 2020, 22, 308. [Google Scholar] [CrossRef]
- Sorensen, A.B.; Sondergaard, M.T.; Overgaard, M.T. Calmodulin in a heartbeat. FEBS J. 2013, 280, 5511–5532. [Google Scholar] [CrossRef]
- Asumda, F.Z.; Chase, P.B. Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells. Differentiation 2012, 83, 106–115. [Google Scholar] [CrossRef]
- Sweeney, H.L.; Brito, R.M.; Rosevear, P.R.; Putkey, J.A. The low-affinity Ca2(+)-binding sites in cardiac/slow skeletal muscle troponin C perform distinct functions: Site I alone cannot trigger contraction. Proc. Natl. Acad. Sci. USA 1990, 87, 9538–9542. [Google Scholar] [CrossRef]
- Hannon, J.D.; Chase, P.B.; Martyn, D.A.; Huntsman, L.L.; Kushmerick, M.J.; Gordon, A.M. Calcium-independent activation of skeletal muscle fibers by a modified form of cardiac troponin C. Biophys. J. 1993, 64, 1632–1637. [Google Scholar] [CrossRef][Green Version]
- Davis, J.P.; Shettigar, V.; Tikunova, S.B.; Little, S.C.; Liu, B.; Siddiqui, J.K.; Janssen, P.M.; Ziolo, M.T.; Walton, S.D. Designing proteins to combat disease: Cardiac troponin C as an example. Arch. Biochem. Biophys. 2016, 601, 4–10. [Google Scholar] [CrossRef]
- Liu, B.; Lee, R.S.; Biesiadecki, B.J.; Tikunova, S.B.; Davis, J.P. Engineered troponin C constructs correct disease-related cardiac myofilament calcium sensitivity. J. Biol. Chem. 2012, 287, 20027–20036. [Google Scholar] [CrossRef]
- Shettigar, V.; Zhang, B.; Little, S.C.; Salhi, H.E.; Hansen, B.J.; Li, N.; Zhang, J.; Roof, S.R.; Ho, H.T.; Brunello, L.; et al. Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease. Nat. Commun. 2016, 7, 10794. [Google Scholar] [CrossRef] [PubMed]
- Tikunova, S.B.; Davis, J.P. Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J. Biol. Chem. 2004, 279, 35341–35352. [Google Scholar] [CrossRef] [PubMed]
- Tikunova, S.B.; Liu, B.; Swindle, N.; Little, S.C.; Gomes, A.V.; Swartz, D.R.; Davis, J.P. Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry 2010, 49, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; George, S.E.; Davis, J.P.; Johnson, J.D. Structural determinants of Ca2+ exchange and affinity in the C terminal of cardiac troponin C. Biochemistry 1998, 37, 14539–14544. [Google Scholar] [CrossRef]
- Tikunova, S.; Belevych, N.; Doan, K.; Reiser, P.J. Desensitizing mouse cardiac troponin C to calcium converts slow muscle towards a fast muscle phenotype. J. Physiol. 2018, 596, 4651–4663. [Google Scholar] [CrossRef]
- Marston, S.; Zamora, J.E. Troponin structure and function: A view of recent progress. J. Muscle Res. Cell Motil. 2020, 41, 71–89. [Google Scholar] [CrossRef]
- Davis, J.P.; Norman, C.; Kobayashi, T.; Solaro, R.J.; Swartz, D.R.; Tikunova, S.B. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys. J. 2007, 92, 3195–3206. [Google Scholar] [CrossRef]
- Norman, C.; Rall, J.A.; Tikunova, S.B.; Davis, J.P. Modulation of the rate of cardiac muscle contraction by troponin C constructs with various calcium binding affinities. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2580–H2587. [Google Scholar] [CrossRef]
- Kreutziger, K.L.; Piroddi, N.; McMichael, J.T.; Tesi, C.; Poggesi, C.; Regnier, M. Calcium binding kinetics of troponin C strongly modulate cooperative activation and tension kinetics in cardiac muscle. J. Mol. Cell Cardiol. 2011, 50, 165–174. [Google Scholar] [CrossRef]
- Mijailovich, S.M.; Prodanovic, M.; Poggesi, C.; Powers, J.D.; Davis, J.; Geeves, M.A.; Regnier, M. The effect of variable troponin C mutation thin filament incorporation on cardiac muscle twitch contractions. J. Mol. Cell Cardiol. 2021, 155, 112–124. [Google Scholar] [CrossRef]
- Biesiadecki, B.J.; Davis, J.P.; Ziolo, M.T.; Janssen, P.M.L. Tri-modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross-bridge cycling kinetics. Biophys. Rev. 2014, 6, 273–289. [Google Scholar] [CrossRef]
- Cass, J.A.; Williams, C.D.; Irving, T.C.; Lauga, E.; Malingen, S.; Daniel, T.L.; Sponberg, S.N. A mechanism for sarcomere breathing: Volume change and advective flow within the myofilament lattice. Biophys. J. 2021, 120, 4079–4090. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.R.; Reynaldo, D.P.; Parvatiyar, M.S.; Dweck, D.; Liang, J.; Jones, M.A.; Sorenson, M.M.; Potter, J.D. Strong cross-bridges potentiate the Ca(2+) affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: A fast kinetic approach. J. Biol. Chem. 2011, 286, 1005–1013. [Google Scholar] [CrossRef]
- Little, S.C.; Biesiadecki, B.J.; Kilic, A.; Higgins, R.S.; Janssen, P.M.; Davis, J.P. The rates of Ca2+ dissociation and cross-bridge detachment from ventricular myofibrils as reported by a fluorescent cardiac troponin C. J. Biol. Chem. 2012, 287, 27930–27940. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Chung, F.; Qu, Y.; Pavlov, D.; Gillis, T.E.; Tikunova, S.B.; Davis, J.P.; Tibbits, G.F. Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol. Genomics 2008, 33, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tikunova, S.B.; Kline, K.P.; Siddiqui, J.K.; Davis, J.P. Disease-related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PLoS ONE 2012, 7, e38259. [Google Scholar] [CrossRef] [PubMed]
- Klass, M.M. Mutation-Specific Calcium Dysregulation in Troponin T Linked Hypertrophic Cardiomyopathy. Ph.D. Thesis, University of Arizona, Tucson, AZ, USA, 2023. [Google Scholar]
- Janssen, P.M.L. Myocardial relaxation in human heart failure: Why sarcomere kinetics should be center-stage. Arch. Biochem. Biophys. 2019, 661, 145–148. [Google Scholar] [CrossRef]
- Siddiqui, J.K.; Tikunova, S.B.; Walton, S.D.; Liu, B.; Meyer, M.; de Tombe, P.P.; Neilson, N.; Kekenes-Huskey, P.M.; Salhi, H.E.; Janssen, P.M.; et al. Myofilament Calcium Sensitivity: Consequences of the Effective Concentration of Troponin I. Front. Physiol. 2016, 7, 632. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Biesiadecki, B.J.; Davis, J.P. Troponin Abnormality and Systolic Function of the Heart. In Troponin: Regulator of Muscle Contraction; Jin, J.-P., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 233–258. [Google Scholar]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca2+ levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2024288118. [Google Scholar] [CrossRef]
- Swartz, D.R.; Moss, R.L.; Greaser, M.L. Calcium alone does not fully activate the thin filament for S1 binding to rigor myofibrils. Biophys. J. 1996, 71, 1891–1904. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reinoso, T.R.; Landim-Vieira, M.; Shi, Y.; Johnston, J.R.; Chase, P.B.; Parvatiyar, M.S.; Landstrom, A.P.; Pinto, J.R.; Tadros, H.J. A comprehensive guide to genetic variants and post-translational modifications of cardiac troponin C. J. Muscle Res. Cell Motil. 2021, 42, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Zot, H.G.; Hasbun, J.E.; Michell, C.A.; Landim-Vieira, M.; Pinto, J.R. Enhanced troponin I binding explains the functional changes produced by the hypertrophic cardiomyopathy mutation A8V of cardiac troponin C. Arch. Biochem. Biophys. 2016, 601, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Saude, E.J.; Wang, X.; Pearlstone, J.R.; Smillie, L.B.; Sykes, B.D. Kinetic studies of calcium and cardiac troponin I peptide binding to human cardiac troponin C using NMR spectroscopy. Eur. Biophys. J. 2002, 31, 245–256. [Google Scholar] [CrossRef]
- Johnston, J.R.; Landim-Vieira, M.; Marques, M.A.; de Oliveira, G.A.P.; Gonzalez-Martinez, D.; Moraes, A.H.; He, H.; Iqbal, A.; Wilnai, Y.; Birk, E.; et al. The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem. 2019, 294, 20054–20069. [Google Scholar] [CrossRef]
- Metzger, J.M.; Moss, R.L. Kinetics of a Ca(2+)-sensitive cross-bridge state transition in skeletal muscle fibers. Effects due to variations in thin filament activation by extraction of troponin C. J. Gen. Physiol. 1991, 98, 233–248. [Google Scholar] [CrossRef]
- Hwang, P.M.; Cai, F.; Pineda-Sanabria, S.E.; Corson, D.C.; Sykes, B.D. The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proc. Natl. Acad. Sci. USA 2014, 111, 14412–14417. [Google Scholar] [CrossRef]
- Finley, N.; Abbott, M.B.; Abusamhadneh, E.; Gaponenko, V.; Dong, W.; Gasmi-Seabrook, G.; Howarth, J.W.; Rance, M.; Solaro, R.J.; Cheung, H.C.; et al. NMR analysis of cardiac troponin C-troponin I complexes: Effects of phosphorylation. FEBS Lett. 1999, 453, 107–112. [Google Scholar] [CrossRef]
- Swindle, N.; Albury, A.N.; Baroud, B.; Burney, M.; Tikunova, S.B. Molecular and functional consequences of mutations in the central helix of cardiac troponin C. Arch. Biochem. Biophys. 2014, 548, 46–53. [Google Scholar] [CrossRef][Green Version]
- Needham, D.M. Machina Carnis: The Biochemistry of Muscular Contraction in Its Historical Development; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Rall, J.A. Mechanism of Muscular Contraction (Perspectives in Physiology); Springer: New York, NY, USA, 2014. [Google Scholar]
- Loong, C.K.; Takeda, A.K.; Badr, M.A.; Rogers, J.S.; Chase, P.B. Slowed Dynamics of Thin Filament Regulatory Units Reduces Ca2+-Sensitivity of Cardiac Biomechanical Function. Cell Mol. Bioeng. 2013, 6, 183–198. [Google Scholar] [CrossRef]
- Solaro, R.J.; Henze, M.; Kobayashi, T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ. Res. 2013, 112, 355–366. [Google Scholar] [CrossRef]
- Solaro, R.J.; Kobayashi, T. Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 2011, 286, 9935–9940. [Google Scholar] [CrossRef] [PubMed]
- Solaro, R.J.; van der Velden, J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J. Mol. Cell Cardiol. 2010, 48, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Nixon, B.R.; Thawornkaiwong, A.; Jin, J.; Brundage, E.A.; Little, S.C.; Davis, J.P.; Solaro, R.J.; Biesiadecki, B.J. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J. Biol. Chem. 2012, 287, 19136–19147. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lopez, J.J.; Biesiadecki, B.J.; Davis, J.P. Protein kinase C phosphomimetics alter thin filament Ca2+ binding properties. PLoS ONE 2014, 9, e86279. [Google Scholar] [CrossRef] [PubMed]
- Nixon, B.R.; Walton, S.D.; Zhang, B.; Brundage, E.A.; Little, S.C.; Ziolo, M.T.; Davis, J.P.; Biesiadecki, B.J. Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment. J. Mol. Cell Cardiol. 2014, 72, 177–185. [Google Scholar] [CrossRef]
- Farman, G.P.; Rynkiewicz, M.J.; Orzechowski, M.; Lehman, W.; Moore, J.R. HCM and DCM cardiomyopathy-linked alpha-tropomyosin mutations influence off-state stability and crossbridge interaction on thin filaments. Arch. Biochem. Biophys. 2018, 647, 84–92. [Google Scholar] [CrossRef]
- Rajan, S.; Ahmed, R.P.; Jagatheesan, G.; Petrashevskaya, N.; Boivin, G.P.; Urboniene, D.; Arteaga, G.M.; Wolska, B.M.; Solaro, R.J.; Liggett, S.B.; et al. Dilated cardiomyopathy mutant tropomyosin mice develop cardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ. Res. 2007, 101, 205–214. [Google Scholar] [CrossRef]
- Loong, C.K.; Badr, M.A.; Chase, P.B. Tropomyosin flexural rigidity and single Ca(2+) regulatory unit dynamics: Implications for cooperative regulation of cardiac muscle contraction and cardiomyocyte hypertrophy. Front. Physiol. 2012, 3, 80. [Google Scholar] [CrossRef]
- Loong, C.K.; Zhou, H.X.; Chase, P.B. Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac alpha-tropomyosin. FEBS Lett. 2012, 586, 3503–3507. [Google Scholar] [CrossRef]
- Wang, F.; Brunet, N.M.; Grubich, J.R.; Bienkiewicz, E.A.; Asbury, T.M.; Compton, L.A.; Mihajlovic, G.; Miller, V.F.; Chase, P.B. Facilitated cross-bridge interactions with thin filaments by familial hypertrophic cardiomyopathy mutations in alpha-tropomyosin. J. Biomed. Biotechnol. 2011, 2011, 435271. [Google Scholar] [CrossRef] [PubMed]
- Day, S.M.; Tardiff, J.C.; Ostap, E.M. Myosin modulators: Emerging approaches for the treatment of cardiomyopathies and heart failure. J. Clin. Investig. 2022, 132, e148557. [Google Scholar] [CrossRef] [PubMed]
- Salhi, H.E.; Hassel, N.C.; Siddiqui, J.K.; Brundage, E.A.; Ziolo, M.T.; Janssen, P.M.; Davis, J.P.; Biesiadecki, B.J. Myofilament Calcium Sensitivity: Mechanistic Insight into TnI Ser-23/24 and Ser-150 Phosphorylation Integration. Front. Physiol. 2016, 7, 567. [Google Scholar] [CrossRef]
- Rayani, K.; Hantz, E.R.; Haji-Ghassemi, O.; Li, A.Y.; Spuches, A.M.; Van Petegem, F.; Solaro, R.J.; Lindert, S.; Tibbits, G.F. The effect of Mg2+ on Ca2+ binding to cardiac troponin C in hypertrophic cardiomyopathy associated TNNC1 variants. FEBS J. 2022, 289, 7446–7465. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.R.; Chase, P.B.; Pinto, J.R. Troponin through the looking-glass: Emerging roles beyond regulation of striated muscle contraction. Oncotarget 2018, 9, 1461–1482. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, V.; Soltisz, A.M.; Moise, N.; Sakuta, G.; Orengo, B.H.; Janssen, P.M.L.; Weinberg, S.H.; Davis, J.P.; Veeraraghavan, R.; Gyorke, S. Distributed synthesis of sarcolemmal and sarcoplasmic reticulum membrane proteins in cardiac myocytes. Basic. Res. Cardiol. 2021, 116, 63. [Google Scholar] [CrossRef]
- Ferrantini, C.; Coppini, R.; Pioner, J.M.; Gentile, F.; Tosi, B.; Mazzoni, L.; Scellini, B.; Piroddi, N.; Laurino, A.; Santini, L.; et al. Pathogenesis of Hypertrophic Cardiomyopathy is Mutation Rather Than Disease Specific: A Comparison of the Cardiac Troponin T E163R and R92Q Mouse Models. J. Am. Heart Assoc. 2017, 6, e005407. [Google Scholar] [CrossRef]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [CrossRef]
- Venkataraman, R.; Baldo, M.P.; Hwang, H.S.; Veltri, T.; Pinto, J.R.; Baudenbacher, F.J.; Knollmann, B.C. Myofilament calcium de-sensitization and contractile uncoupling prevent pause-triggered ventricular tachycardia in mouse hearts with chronic myocardial infarction. J. Mol. Cell Cardiol. 2013, 60, 8–15. [Google Scholar] [CrossRef]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Kirchhof, P.; Sirenko, S.G.; Degen, H.; Greene, A.E.; Schober, T.; Mackow, J.C.; Fabritz, L.; Potter, J.D.; Morad, M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ. Res. 2003, 92, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Pinto, J.R. Suppression of lusitropy as a disease mechanism in cardiomyopathies. Front. Cardiovasc. Med. 2022, 9, 1080965. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Schmidt-Traub, H.; Perrot, A.; Osterziel, K.J.; Gessner, R. First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum. Mutat. 2001, 17, 524. [Google Scholar] [CrossRef]
- Tadros, H.J.; Life, C.S.; Garcia, G.; Pirozzi, E.; Jones, E.G.; Datta, S.; Parvatiyar, M.S.; Chase, P.B.; Allen, H.D.; Kim, J.J.; et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J. Mol. Cell Cardiol. 2020, 142, 118–125. [Google Scholar] [CrossRef]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- Kalyva, A.; Parthenakis, F.I.; Marketou, M.E.; Kontaraki, J.E.; Vardas, P.E. Biochemical characterisation of Troponin C mutations causing hypertrophic and dilated cardiomyopathies. J. Muscle Res. Cell Motil. 2014, 35, 161–178. [Google Scholar] [CrossRef]
- Hassoun, R.; Budde, H.; Mannherz, H.G.; Lodi, M.; Fujita-Becker, S.; Laser, K.T.; Gartner, A.; Klingel, K.; Mohner, D.; Stehle, R.; et al. De Novo Missense Mutations in TNNC1 and TNNI3 Causing Severe Infantile Cardiomyopathy Affect Myofilament Structure and Function and Are Modulated by Troponin Targeting Agents. Int. J. Mol. Sci. 2021, 22, 9625. [Google Scholar] [CrossRef]
- Marques, M.A.; Landim-Vieira, M.; Moraes, A.H.; Sun, B.; Johnston, J.R.; Dieseldorff Jones, K.M.; Cino, E.A.; Parvatiyar, M.S.; Valera, I.C.; Silva, J.L.; et al. Anomalous structural dynamics of minimally frustrated residues in cardiac troponin C triggers hypertrophic cardiomyopathy. Chem. Sci. 2021, 12, 7308–7323. [Google Scholar] [CrossRef]
- Martins, A.S.; Parvatiyar, M.S.; Feng, H.Z.; Bos, J.M.; Gonzalez-Martinez, D.; Vukmirovic, M.; Turna, R.S.; Sanchez-Gonzalez, M.A.; Badger, C.D.; Zorio, D.A.R.; et al. In Vivo Analysis of Troponin C Knock-In (A8V) Mice: Evidence that TNNC1 Is a Hypertrophic Cardiomyopathy Susceptibility Gene. Circ. Cardiovasc. Genet. 2015, 8, 653–664. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, J.; Stienen, G.J.M. Cardiac Disorders and Pathophysiology of Sarcomeric Proteins. Physiol. Rev. 2019, 99, 381–426. [Google Scholar] [CrossRef]
- Willott, R.H.; Gomes, A.V.; Chang, A.N.; Parvatiyar, M.S.; Pinto, J.R.; Potter, J.D. Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J. Mol. Cell Cardiol. 2010, 48, 882–892. [Google Scholar] [CrossRef]
- Sayle, R.A.; Milner-White, E.J. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 1995, 20, 374. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Parvatiyar, M.S.; Pinto, J.R.; Marquardt, M.L.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Potter, J.D.; Ackerman, M.J. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell Cardiol. 2008, 45, 281–288. [Google Scholar] [CrossRef]
- Pinto, J.R.; Parvatiyar, M.S.; Jones, M.A.; Liang, J.; Ackerman, M.J.; Potter, J.D. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 2009, 284, 19090–19100. [Google Scholar] [CrossRef]
- Albury, A.N.; Swindle, N.; Swartz, D.R.; Tikunova, S.B. Effect of hypertrophic cardiomyopathy-linked troponin C mutations on the response of reconstituted thin filaments to calcium upon troponin I phosphorylation. Biochemistry 2012, 51, 3614–3621. [Google Scholar] [CrossRef]
- Li, A.Y.; Stevens, C.M.; Liang, B.; Rayani, K.; Little, S.; Davis, J.; Tibbits, G.F. Familial hypertrophic cardiomyopathy related cardiac troponin C L29Q mutation alters length-dependent activation and functional effects of phosphomimetic troponin I*. PLoS ONE 2013, 8, e79363. [Google Scholar] [CrossRef] [PubMed]
- Dweck, D.; Hus, N.; Potter, J.D. Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J. Biol. Chem. 2008, 283, 33119–33128. [Google Scholar] [CrossRef] [PubMed]
- Neulen, A.; Stehle, R.; Pfitzer, G. The cardiac troponin C mutation Leu29Gln found in a patient with hypertrophic cardiomyopathy does not alter contractile parameters in skinned murine myocardium. Basic. Res. Cardiol. 2009, 104, 751–760. [Google Scholar] [CrossRef]
- Gollapudi, S.K.; Chandra, M. Cardiomyopathy-Related Mutations in Cardiac Troponin C, L29Q and G159D, Have Divergent Effects on Rat Cardiac Myofiber Contractile Dynamics. Biochem. Res. Int. 2012, 2012, 824068. [Google Scholar] [CrossRef]
- Parvatiyar, M.S.; Landstrom, A.P.; Figueiredo-Freitas, C.; Potter, J.D.; Ackerman, M.J.; Pinto, J.R. A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 2012, 287, 31845–31855. [Google Scholar] [CrossRef]
- Swindle, N.; Tikunova, S.B. Hypertrophic cardiomyopathy-linked mutation D145E drastically alters calcium binding by the C-domain of cardiac troponin C. Biochemistry 2010, 49, 4813–4820. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.R.; Siegfried, J.D.; Parvatiyar, M.S.; Li, D.; Norton, N.; Jones, M.A.; Liang, J.; Potter, J.D.; Hershberger, R.E. Functional characterization of TNNC1 rare variants identified in dilated cardiomyopathy. J. Biol. Chem. 2011, 286, 34404–34412. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef]
- Lu, Q.W.; Wu, X.Y.; Morimoto, S. Inherited cardiomyopathies caused by troponin mutations. J. Geriatr. Cardiol. 2013, 10, 91–101. [Google Scholar]
- van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; van Veldhuisen, D.J.; van der Werf, R.; Jongbloed, J.D.; Paulus, W.J.; Dooijes, D.; van den Berg, M.P. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 2010, 121, 2169–2175. [Google Scholar] [CrossRef]
- Lim, C.C.; Yang, H.; Yang, M.; Wang, C.K.; Shi, J.; Berg, E.A.; Pimentel, D.R.; Gwathmey, J.K.; Hajjar, R.J.; Helmes, M.; et al. A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomyocyte contractility. Biophys. J. 2008, 94, 3577–3589. [Google Scholar] [CrossRef]
- Dweck, D.; Reynaldo, D.P.; Pinto, J.R.; Potter, J.D. A dilated cardiomyopathy troponin C mutation lowers contractile force by reducing strong myosin-actin binding. J. Biol. Chem. 2010, 285, 17371–17379. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Truszkowska, G.; Chmielewski, P.; Rydzanicz, M.; Kosinska, J.; Rywik, T.; Biernacka, A.; Spiewak, M.; Kostrzewa, G.; Stepien-Wojno, M.; et al. Analysis of De Novo Mutations in Sporadic Cardiomyopathies Emphasizes Their Clinical Relevance and Points to Novel Candidate Genes. J. Clin. Med. 2020, 9, 370. [Google Scholar] [CrossRef]
- Landim-Vieira, M.; Johnston, J.R.; Ji, W.; Mis, E.K.; Tijerino, J.; Spencer-Manzon, M.; Jeffries, L.; Hall, E.K.; Panisello-Manterola, D.; Khokha, M.K.; et al. Familial Dilated Cardiomyopathy Associated With a Novel Combination of Compound Heterozygous TNNC1 Variants. Front. Physiol. 2019, 10, 1612. [Google Scholar] [CrossRef]
- Mogensen, J.; Murphy, R.T.; Shaw, T.; Bahl, A.; Redwood, C.; Watkins, H.; Burke, M.; Elliott, P.M.; McKenna, W.J. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2033–2040. [Google Scholar] [CrossRef]
- Kaski, J.P.; Burch, M.; Elliott, P.M. Mutations in the cardiac Troponin C gene are a cause of idiopathic dilated cardiomyopathy in childhood. Cardiol. Young 2007, 17, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Biesiadecki, B.J.; Kobayashi, T.; Walker, J.S.; Solaro, R.J.; de Tombe, P.P. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 2007, 100, 1486–1493. [Google Scholar] [CrossRef]
- Robinson, P.; Griffiths, P.J.; Watkins, H.; Redwood, C.S. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 2007, 101, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Dyer, E.C.; Jacques, A.M.; Hoskins, A.C.; Ward, D.G.; Gallon, C.E.; Messer, A.E.; Kaski, J.P.; Burch, M.; Kentish, J.C.; Marston, S.B. Functional analysis of a unique troponin c mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail. 2009, 2, 456–464. [Google Scholar] [CrossRef]
- Yadav, S.; Sitbon, Y.H.; Kazmierczak, K.; Szczesna-Cordary, D. Hereditary heart disease: Pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains. Pflugers Arch. 2019, 471, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, C.; Gomez-Guerrero, C.; Martin-Ventura, J.L.; Blanco-Colio, L.; Lavin, B.; Mallavia, B.; Tarin, C.; Mas, S.; Ortiz, A.; Egido, J. Animal models of cardiovascular diseases. J. Biomed. Biotechnol. 2011, 2011, 497841. [Google Scholar] [CrossRef]
- Camacho, P.; Fan, H.; Liu, Z.; He, J.Q. Small mammalian animal models of heart disease. Am. J. Cardiovasc. Dis. 2016, 6, 70–80. [Google Scholar]
- Kawai, M.; Johnston, J.R.; Karam, T.; Wang, L.; Singh, R.K.; Pinto, J.R. Myosin Rod Hypophosphorylation and CB Kinetics in Papillary Muscles from a TnC-A8V KI Mouse Model. Biophys. J. 2017, 112, 1726–1736. [Google Scholar] [CrossRef]
- Gonzalez-Martinez, D.; Johnston, J.R.; Landim-Vieira, M.; Ma, W.; Antipova, O.; Awan, O.; Irving, T.C.; Bryant Chase, P.; Pinto, J.R. Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell Cardiol. 2018, 123, 26–37. [Google Scholar] [CrossRef]
- Dieseldorff Jones, K.M.; Vied, C.; Valera, I.C.; Chase, P.B.; Parvatiyar, M.S.; Pinto, J.R. Sexual dimorphism in cardiac transcriptome associated with a troponin C murine model of hypertrophic cardiomyopathy. Physiol. Rep. 2020, 8, e14396. [Google Scholar] [CrossRef]
- Feest, E.R.; Steven Korte, F.; Tu, A.Y.; Dai, J.; Razumova, M.V.; Murry, C.E.; Regnier, M. Thin filament incorporation of an engineered cardiac troponin C variant (L48Q) enhances contractility in intact cardiomyocytes from healthy and infarcted hearts. J. Mol. Cell Cardiol. 2014, 72, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.D.; Kooiker, K.B.; Mason, A.B.; Teitgen, A.E.; Flint, G.V.; Tardiff, J.C.; Schwartz, S.D.; McCulloch, A.D.; Regnier, M.; Davis, J.; et al. Modulating the tension-time integral of the cardiac twitch prevents dilated cardiomyopathy in murine hearts. JCI Insight 2020, 5, e142446. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.K.; Singh, S.; Fan, Q.; Hernandez, A.; Portillo, J.P.; Reiser, P.J.; Tikunova, S.B. Knock-in mice harboring a Ca(2+) desensitizing mutation in cardiac troponin C develop early onset dilated cardiomyopathy. Front. Physiol. 2015, 6, 242. [Google Scholar] [CrossRef]
- Tikunova, S.B.; Davis, J.P.; Rall, J.A. Engineering cardiac troponin C (cTnC) mutants with dramatically altered Ca2+ dissociation rates as molecular tools to study cardiac muscle relaxation. Biophys. J. 2004, 86, 394A. [Google Scholar]
- Veltri, T.; Landim-Vieira, M.; Parvatiyar, M.S.; Gonzalez-Martinez, D.; Dieseldorff Jones, K.M.; Michell, C.A.; Dweck, D.; Landstrom, A.P.; Chase, P.B.; Pinto, J.R. Hypertrophic Cardiomyopathy Cardiac Troponin C Mutations Differentially Affect Slow Skeletal and Cardiac Muscle Regulation. Front. Physiol. 2017, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Coldren, W.H.; Tikunova, S.B.; Davis, J.P.; Lindert, S. Discovery of Novel Small-Molecule Calcium Sensitizers for Cardiac Troponin C: A Combined Virtual and Experimental Screening Approach. J. Chem. Inf. Model. 2020, 60, 3648–3661. [Google Scholar] [CrossRef]
- Hantz, E.R.; Tikunova, S.B.; Belevych, N.; Davis, J.P.; Reiser, P.J.; Lindert, S. Targeting Troponin C with Small Molecules Containing Diphenyl Moieties: Calcium Sensitivity Effects on Striated Muscles and Structure-Activity Relationship. J. Chem. Inf. Model. 2023, 63, 3462–3473. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikunova, S.B.; Thuma, J.; Davis, J.P. Mouse Models of Cardiomyopathies Caused by Mutations in Troponin C. Int. J. Mol. Sci. 2023, 24, 12349. https://doi.org/10.3390/ijms241512349
Tikunova SB, Thuma J, Davis JP. Mouse Models of Cardiomyopathies Caused by Mutations in Troponin C. International Journal of Molecular Sciences. 2023; 24(15):12349. https://doi.org/10.3390/ijms241512349
Chicago/Turabian StyleTikunova, Svetlana B., Jenna Thuma, and Jonathan P. Davis. 2023. "Mouse Models of Cardiomyopathies Caused by Mutations in Troponin C" International Journal of Molecular Sciences 24, no. 15: 12349. https://doi.org/10.3390/ijms241512349
APA StyleTikunova, S. B., Thuma, J., & Davis, J. P. (2023). Mouse Models of Cardiomyopathies Caused by Mutations in Troponin C. International Journal of Molecular Sciences, 24(15), 12349. https://doi.org/10.3390/ijms241512349