
Dihydroxyphenylacetaldehyde Lowering Treatment Improves Locomotor and Neurochemical Abnormalities in the Rat Rotenone Model: Relevance to the Catecholaldehyde Hypothesis for the Pathogenesis of Parkinson’s Disease

 ,
, 
Abstract
1. Introduction
The Molecular Basis of the Catecholaldehyde Hypothesis
2. Results
2.1. Rotenone Induces Neurochemical and Functional Abnormlities
2.2. Deprrenyl Alone or with NAC Mitigate the Rotenone-Induced Neurochemical and Functional Abnormlities
3. Discussion
3.1. The Centrality of DOPAL in Dopamine Neuronal Toxicity
3.2. Should MAOI for Parkinson’s Disease Be Revisited?
3.3. Study’s Limitations and Strengths
4. Material and Methods
4.1. Animals
4.2. Materials
4.3. Study Protocol
4.4. Locomotor Tests
4.5. Neurochemical Assays
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Mash, D.C.; Kopin, I.J.; Sharabi, Y. Determinants of denervation-independent depletion of putamen dopamine in Parkinson’s disease and multiple system atrophy. Park. Relat. Disord. 2017, 35, 88–91. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Holmes, C.; Sullivan, P.; Mash, D.C.; Sidransky, E.; Stefani, A.; Kopin, I.J.; Sharabi, Y. Deficient vesicular storage: A common theme in catecholaminergic neurodegeneration. Park. Relat. Disord. 2015, 21, 1013–1022. [Google Scholar] [CrossRef]
- Panneton, W.M.; Kumar, V.B.; Gan, Q.; Burke, W.J.; Galvin, J.E. The neurotoxicity of DOPAL: Behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS ONE 2010, 5, e15251. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.J.; Li, S.W.; Williams, E.A.; Nonneman, R.; Zahm, D.S. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: Implications for Parkinson’s disease pathogenesis. Brain Res. 2003, 989, 205–213. [Google Scholar] [CrossRef]
- Mattammal, M.B.; Haring, J.H.; Chung, H.D.; Raghu, G.; Strong, R. An endogenous dopaminergic neurotoxin: Implication for Parkinson’s disease. Neurodegeneration 1995, 4, 271–281. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Miller, G.W.; Alter, S.; Strong, R.; Mash, D.C.; Kopin, I.J.; Sharabi, Y. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J. Neurochem. 2013, 126, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.W.; Erickson, J.D.; Perez, J.T.; Penland, S.N.; Mash, D.C.; Rye, D.B.; Levey, A.I. Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson’s disease. Exp. Neurol. 1999, 156, 138–148. [Google Scholar] [CrossRef]
- Okamura, N.; Villemagne, V.L.; Drago, J.; Pejoska, S.; Dhamija, R.K.; Mulligan, R.S.; Ellis, J.R.; Ackermann, U.; O’Keefe, G.; Jones, G.; et al. In vivo measurement of vesicular monoamine transporter type 2 density in Parkinson disease with (18)F-AV-133. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 223–228. [Google Scholar]
- Fitzmaurice, A.G.; Rhodes, S.L.; Lulla, A.; Murphy, N.P.; Lam, H.A.; O’Donnell, K.C.; Barnhill, L.; Casida, J.E.; Cockburn, M.; Sagasti, A.; et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. USA 2013, 110, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, L.M.; Florang, V.R.; Doorn, J.A. Catechol and aldehyde moieties of 3,4-dihydroxyphenylacetaldehyde contribute to tyrosine hydroxylase inhibition and neurotoxicity. Brain Res. 2012, 1474, 100–109. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sharabi, Y.; Sullivan, P.; Isonaka, R.; Goldstein, D.S. 3,4-Dihydroxyphenylacetaldehyde-Induced Protein Modifications and Their Mitigation by N-Acetylcysteine. J. Pharmacol. Exp. Ther. 2018, 366, 113–124. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Pekker, M.J.; Eisenhofer, G.; Sharabi, Y. Computational modeling reveals multiple abnormalities of myocardial noradrenergic function in Lewy body diseases. JCI Insight 2019, 4, e130441. [Google Scholar] [CrossRef]
- Wimalasena, K. Vesicular monoamine transporters: Structure-function, pharmacology, and medicinal chemistry. Med. Res. Rev. 2011, 31, 483–519. [Google Scholar] [CrossRef]
- Takahashi, N.; Miner, L.L.; Sora, I.; Ujike, H.; Revay, R.S.; Kostic, V.; Jackson-Lewis, V.; Przedborski, S.; Uhl, G.R. VMAT2 knockout mice: Heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc. Natl. Acad. Sci. USA 1997, 94, 9938–9943. [Google Scholar] [CrossRef]
- Taylor, T.N.; Caudle, W.M.; Miller, G.W. VMAT2-Deficient Mice Display Nigral and Extranigral Pathology and Motor and Nonmotor Symptoms of Parkinson’s Disease. Park. Dis. 2011, 2011, 124165. [Google Scholar] [CrossRef]
- Alter, S.P.; Lenzi, G.M.; Bernstein, A.I.; Miller, G.W. Vesicular integrity in Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2013, 13, 362. [Google Scholar] [CrossRef] [PubMed]
- Deza-Ponzio, R.; Herrera, M.L.; Bellini, M.J.; Virgolini, M.B.; Herenu, C.B. Aldehyde dehydrogenase 2 in the spotlight: The link between mitochondria and neurodegeneration. Neurotoxicology 2018, 68, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Grunblatt, E.; Riederer, P. Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. J. Neural Transm. 2016, 123, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Joshi, A.U.; Mochly-Rosen, D. The Role of Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) in Neuropathology and Neurodegeneration. Acta Neurol. Taiwanica 2016, 25, 111–123. [Google Scholar]
- Wey, M.C.; Fernandez, E.; Martinez, P.A.; Sullivan, P.; Goldstein, D.S.; Strong, R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: Implications for Parkinson’s disease. PLoS ONE 2012, 7, e31522. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.N.; Zhang, J.S.; Xiang, J.; Yu, Z.H.; Zhang, W.; Cai, M.; Li, X.T.; Wu, T.; Li, W.W.; Cai, D.F. Subcutaneous rotenone rat model of Parkinson’s disease: Dose exploration study. Brain Res. 2017, 1655, 104–113. [Google Scholar] [CrossRef]
- Landau, R.; Halperin, R.; Sullivan, P.; Zibly, Z.; Leibowitz, A.; Goldstein, D.S.; Sharabi, Y. The rat rotenone model reproduces the abnormal pattern of central catecholamine metabolism found in Parkinson’s disease. Dis. Models Mech. 2022, 15, dmm049082. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Sharabi, Y. Comparison of Monoamine Oxidase Inhibitors in Decreasing Production of the Autotoxic Dopamine Metabolite 3,4-Dihydroxyphenylacetaldehyde in PC12 Cells. J. Pharmacol. Exp. Ther. 2016, 356, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Youdim, M.B. Monoamine oxidase activity and monoamine metabolism in brains of parkinsonian patients treated with l-deprenyl. J. Neurochem. 1986, 46, 1359–1365. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Pekker, M.J.; Sullivan, P.; Isonaka, R.; Sharabi, Y. Modeling the Progression of Cardiac Catecholamine Deficiency in Lewy Body Diseases. J. Am. Heart Assoc. 2022, 11, e024411. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Sharabi, Y. N-Acetylcysteine Prevents the Increase in Spontaneous Oxidation of Dopamine During Monoamine Oxidase Inhibition in PC12 Cells. Neurochem. Res. 2017, 42, 3289–3295. [Google Scholar] [CrossRef]
- Anton, S.D. Can non-nutritive sweeteners enhance outcomes of weight loss interventions? Obesity 2014, 22, 1413–1414. [Google Scholar] [CrossRef]
- Goldstein, D.S. The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know. Int. J. Mol. Sci. 2021, 22, 5999. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef]
- Wahdan, S.A.; Tadros, M.G.; Khalifa, A.E. Antioxidant and antiapoptotic actions of selegiline protect against 3-NP-induced neurotoxicity in rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 905–917. [Google Scholar] [CrossRef]
- Carmo-Goncalves, P.; Coelho-Cerqueira, E.; de Araujo Lima, V.; Follmer, C. Alpha-synuclein in Parkinson’s disease: A villain or tragic hero? A critical view of the formation of alpha-synuclein aggregates induced by dopamine metabolites and viral infection. Expert Rev. Neurother. 2023, 23, 321–330. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Isonaka, R.; Sharabi, Y.; Goldstein, D.S. 3,4-Dihydroxyphenylacetaldehyde Is More Efficient than Dopamine in Oligomerizing and Quinonizing alpha-Synuclein. J. Pharmacol. Exp. Ther. 2020, 372, 157–165. [Google Scholar] [CrossRef]
- Kumar, V.B.; Hsu, F.F.; Lakshmi, V.M.; Gillespie, K.N.; Burke, W.J. Aldehyde adducts inhibit 3,4-dihydroxyphenylacetaldehyde-induced alpha-synuclein aggregation and toxicity: Implication for Parkinson neuroprotective therapy. Eur. J. Pharmacol. 2019, 845, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lima, V.A.; do Nascimento, L.A.; Eliezer, D.; Follmer, C. Role of Parkinson’s Disease-Linked Mutations and N-Terminal Acetylation on the Oligomerization of alpha-Synuclein Induced by 3,4-Dihydroxyphenylacetaldehyde. ACS Chem. Neurosci. 2019, 10, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Yacoub, A.; Kunz, A.; Aranki, B.; Serobyan, G.; Cohn, W.; Whitelegge, J.P.; Watson, J.B. Enhanced mitochondrial inhibition by 3,4-dihydroxyphenyl-acetaldehyde (DOPAL)-oligomerized alpha-synuclein. J. Neurosci. Res. 2019, 97, 1689–1705. [Google Scholar] [PubMed]
- Li, S.W.; Lin, T.S.; Minteer, S.; Burke, W.J. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: Possible role in Parkinson’s disease pathogenesis. Mol. Brain Res. 2001, 93, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kristal, B.S.; Conway, A.D.; Brown, A.M.; Jain, J.C.; Ulluci, P.A.; Li, S.W.; Burke, W.J. Selective dopaminergic vulnerability: 3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free Radic. Biol. Med. 2001, 30, 924–931. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Terrin, F.; Sandre, M.; Faustini, G.; Thor, A.; Adams, S.; Berti, G.; Cogo, S.; De Lazzari, F.; et al. DOPAL initiates alphaSynuclein-dependent impaired proteostasis and degeneration of neuronal projections in Parkinson’s disease. NPJ Park. Dis. 2023, 9, 42. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate alpha-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J. Neural Transm. 2020, 127, 131–147. [Google Scholar] [CrossRef]
- Martinez, P.A.; Martinez, V.E.; Rani, S.; Murrell, M.; Javors, M.; Gelfond, J.; Doorn, J.A.; Fernandez, E.; Strong, R. Impaired aldehyde detoxification exacerbates motor deficits in an alpha-synuclein mouse model of Parkinson’s disease. Brain Behav. 2023, e3150. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Mercatelli, D.; Novello, S.; Arcuri, L.; Brugnoli, A.; Vincenzi, F.; Russo, I.; Berti, G.; Mabrouk, O.S.; Kennedy, R.T.; et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Stednitz, S.J.; Freshner, B.; Shelton, S.; Shen, T.; Black, D.; Gahtan, E. Selective toxicity of L-DOPA to dopamine transporter-expressing neurons and locomotor behavior in zebrafish larvae. Neurotoxicol. Teratol. 2015, 52 Pt A, 51–56. [Google Scholar] [CrossRef]
- Badillo-Ramirez, I.; Saniger, J.M.; Rivas-Arancibia, S. 5-S-cysteinyl-dopamine, a neurotoxic endogenous metabolite of dopamine: Implications for Parkinson’s disease. Neurochem. Int. 2019, 129, 104514. [Google Scholar] [CrossRef]
- Herrera, A.; Munoz, P.; Steinbusch HW, M.; Segura-Aguilar, J. Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Deus, C.M.; Teixeira, J.; Raimundo, N.; Tucci, P.; Borges, F.; Saso, L.; Oliveira, P.J. Modulation of cellular redox environment as a novel therapeutic strategy for Parkinson’s disease. Eur. J. Clin. Investig. 2022, 52, e13820. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated with Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Parkinson Study Group QE3 Investigators; Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Prasuhn, J.; Bruggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; Konig, I.R.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 31. [Google Scholar] [CrossRef]
- Katz, M.; Won, S.J.; Park, Y.; Orr, A.; Jones, D.P.; Swanson, R.A.; Glass, G.A. Cerebrospinal fluid concentrations of N-acetylcysteine after oral administration in Parkinson’s disease. Park. Relat. Disord. 2015, 21, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Schallert, T.; Keep, R.F.; Wu, J.; Hoff, J.T.; Xi, G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke 2002, 33, 2478–2484. [Google Scholar] [CrossRef]
- Schaar, K.L.; Brenneman, M.M.; Savitz, S.I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2010, 2, 13. [Google Scholar] [CrossRef]
- Holmes, C.; Eisenhofer, G.; Goldstein, D.S. Improved assay for plasma dihydroxyphenylacetic acid and other catechols using high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Appl. 1994, 653, 131–138. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Catechol | VEH | ROT | ROT+DEP | ROT+DEP+NAC |
|---|---|---|---|---|
| DHPG | 50.37 ± 3.96 abc | 96.17 ± 5.06 ade | 27.71 ± 4.87 bd | 29.46 ± 2.37 ce |
| NE | 112.90 ± 11.38 bc | 112.03 ± 11.4 de | 284.45 ± 27.81 bdf | 422.02 ± 50.54 cef |
| DOPA | 44.28 ± 3.51 abc | 65.53 ± 4.33 ade | 32.34 ± 1.41 bd | 29.66 ± 1.44 ce |
| DOPAL | 685.94 ± 72.6 abc | 1152.1 ± 97.38 ade | 429.46 ± 96.88 b*df | 192.6 ± 37.93 cef |
| DA | 2665.91 ± 203.7 abc | 1417.14 ± 101.42 ade | 6304.3 ± 482.63 bdf | 4621.73 ± 1078.77 cef |
| DOPAC | 957.93 ± 88.81 c | 1029.42 ± 133.55 e | 754.8 ± 88.80 | 516.14 ± 300.14 ce |
| DOPET | 110.67 ± 10.86 bc | 125.68 ± 26.76 de | 52.09 ± 7.55 bdf | 23.28 ± 17.43 cef |
| CysDA | 25.05 ± 2.33 abc | 13.75 ± 1.61 ad | 48.78 ± 13.04 bd | 53.51 ± 14.28 c |
| Catechol Ratio | Process | VEH | ROT | ROT+DEP | ROT+DEP+NAC |
|---|---|---|---|---|---|
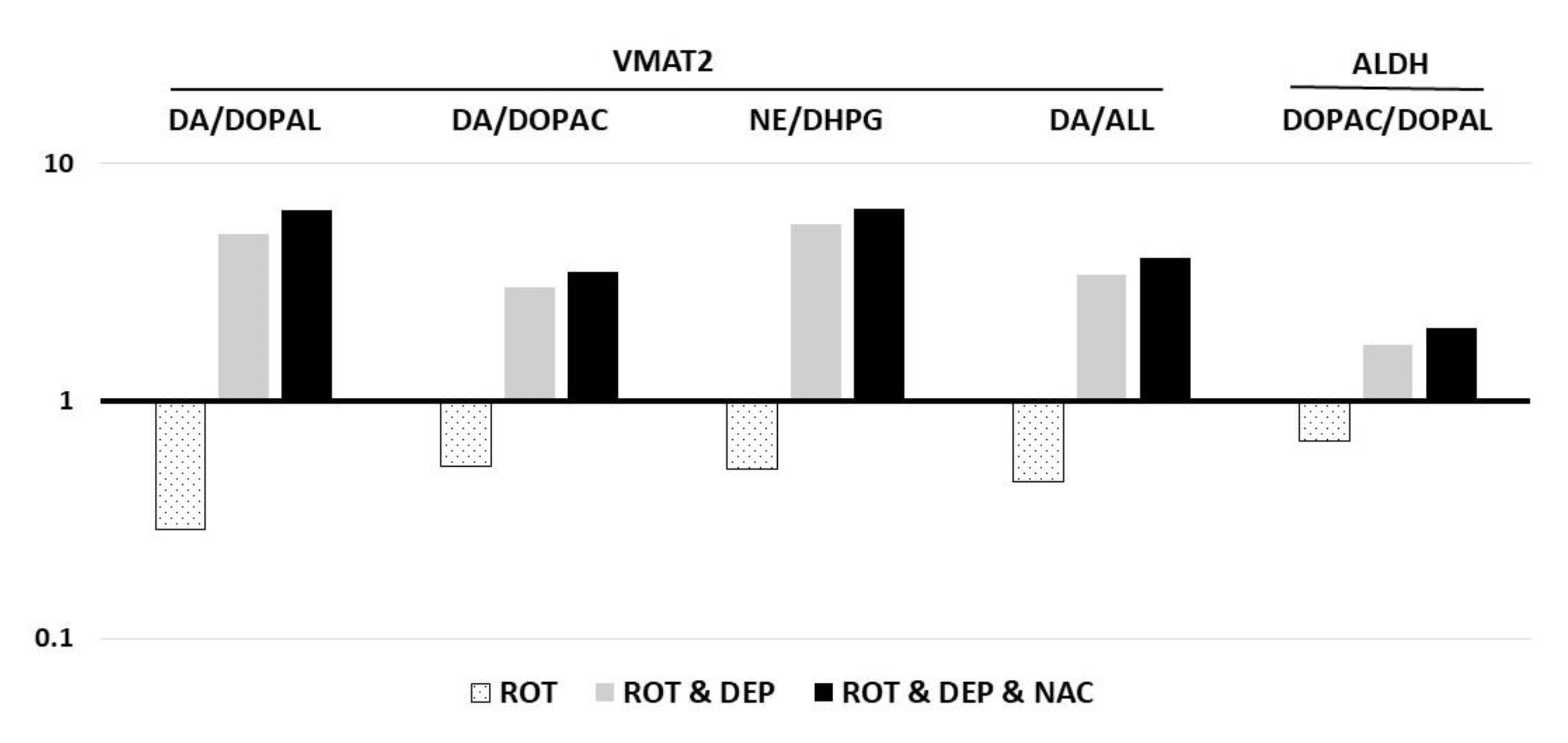
| DA/DOPAL | VMAT2 | 4.42 ± 0.64 abc | 1.27 ± 0.13 ade | 22.27 ± 5.7 bd | 28.22 ± 4.14 ce |
| DA/DOPAC | VMAT2 | 3.01 ± 0.34 abc | 1.6 ± 0.3 ade | 9.11 ± 1.3 bd | 10.5 ± 1.11 ce |
| NE/DHPG | VMAT2 | 2.28 ± 0.20 abc | 1.18 ± 0.12 ade | 12.68 ± 2.46 bd | 14.61 ± 1.43 ce |
| DA/ALL | VMAT2 | 1.72 ± 0.21 ac | 0.79 ± 0.18 ade | 5.89 ± 1.01 d | 6.92 ± 0.54 ce |
| DOPAC/DOPAL | ALDH | 1.43 ± 0.07 abc | 0.98 ± 0.14 ade | 2.48 ± 0.49 bd | 2.89 ± 0.45 ce |
| Motor Parameter | Day | VEH | ROT | ROT+DEP | ROT+DEP+NAC |
|---|---|---|---|---|---|
| Rearing (counts) | BL | 6.50 ± 0.89 | 4.18 ± 0.69 | 6.00 ± 1.20 | 6.00 ± 0.73 |
| End | 3.8 ± 0.53 a | 2.00 ± 0.38 ade | 3.57 ± 0.48 d | 3.57 ± 0.30 e | |
| Distance (cm) | BL | 2962.3 ± 240.8 | 2831.4 ± 250.0 | 1328.0 ± 524.3 | 2325.2 ± 346.6 |
| END | 2291.1 ± 201.9 abc | 972.8 ± 132.7 ade | 1540.5 ± 259.84 bd | 1765.9 ± 67.9 ce | |
| Velocity (cm/min) | BL | 9.87 ± 0.80 | 9.44 ± 0.83 | 4.43 ± 1.75 | 8.31 ± 0.89 |
| END | 7.64 ± 0.68 abc | 3.23 ± 0.32 ade | 5.02 ± 0.85 bd | 5.39 ± 0.39 ce |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khashab, R.; Gutman-Sharabi, N.; Shabtai, Z.; Landau, R.; Halperin, R.; Fay-Karmon, T.; Leibowitz, A.; Sharabi, Y. Dihydroxyphenylacetaldehyde Lowering Treatment Improves Locomotor and Neurochemical Abnormalities in the Rat Rotenone Model: Relevance to the Catecholaldehyde Hypothesis for the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 12522. https://doi.org/10.3390/ijms241512522
Khashab R, Gutman-Sharabi N, Shabtai Z, Landau R, Halperin R, Fay-Karmon T, Leibowitz A, Sharabi Y. Dihydroxyphenylacetaldehyde Lowering Treatment Improves Locomotor and Neurochemical Abnormalities in the Rat Rotenone Model: Relevance to the Catecholaldehyde Hypothesis for the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(15):12522. https://doi.org/10.3390/ijms241512522
Chicago/Turabian StyleKhashab, Rawan, Naama Gutman-Sharabi, Zehava Shabtai, Regev Landau, Reut Halperin, Tsviya Fay-Karmon, Avshalom Leibowitz, and Yehonatan Sharabi. 2023. "Dihydroxyphenylacetaldehyde Lowering Treatment Improves Locomotor and Neurochemical Abnormalities in the Rat Rotenone Model: Relevance to the Catecholaldehyde Hypothesis for the Pathogenesis of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 15: 12522. https://doi.org/10.3390/ijms241512522
APA StyleKhashab, R., Gutman-Sharabi, N., Shabtai, Z., Landau, R., Halperin, R., Fay-Karmon, T., Leibowitz, A., & Sharabi, Y. (2023). Dihydroxyphenylacetaldehyde Lowering Treatment Improves Locomotor and Neurochemical Abnormalities in the Rat Rotenone Model: Relevance to the Catecholaldehyde Hypothesis for the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences, 24(15), 12522. https://doi.org/10.3390/ijms241512522