
Repurposing of World-Approved Drugs for Potential Inhibition against Human Carbonic Anhydrase I: A Computational Study


Abstract
:1. Introduction
2. Results and Discussion
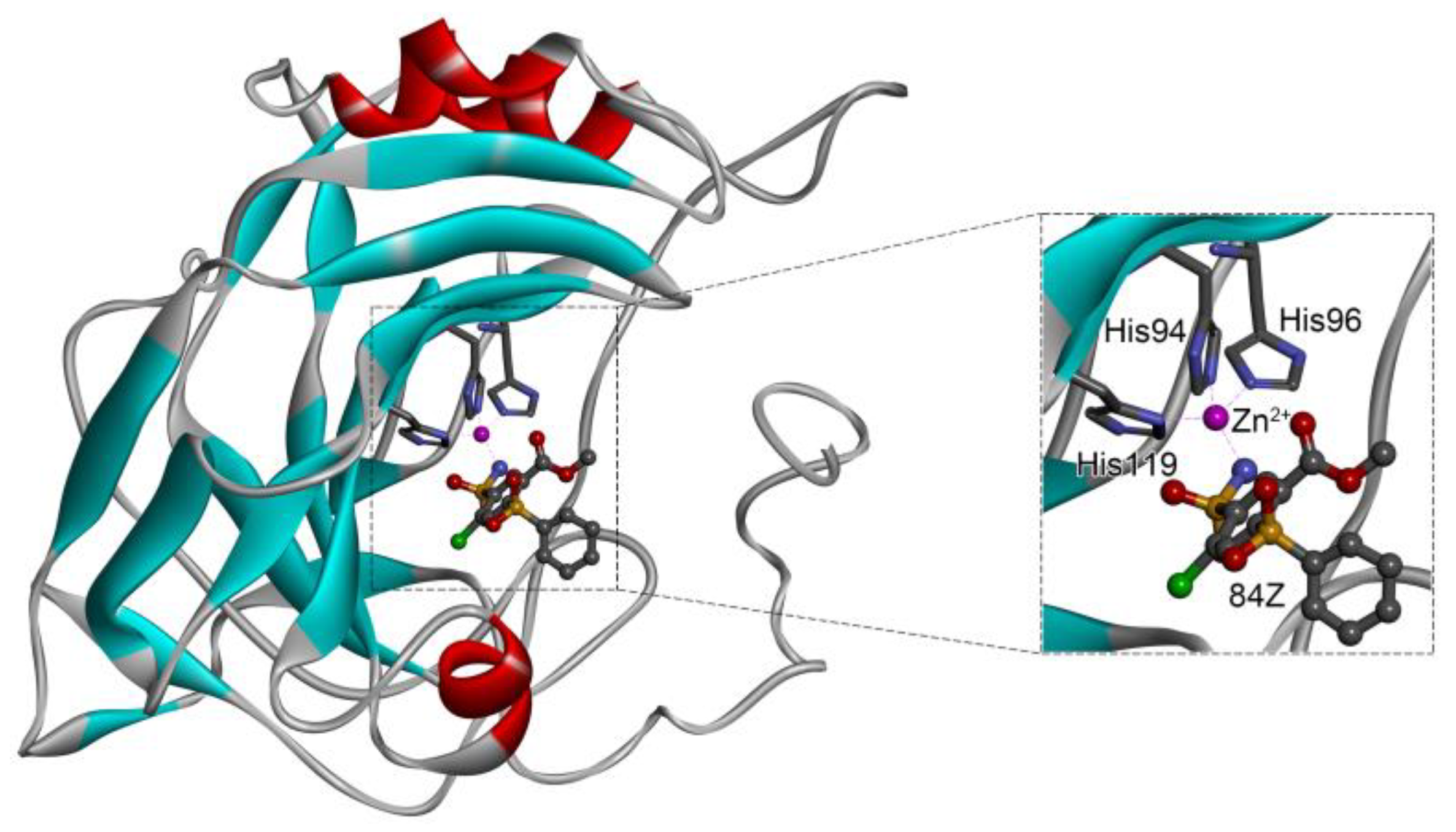
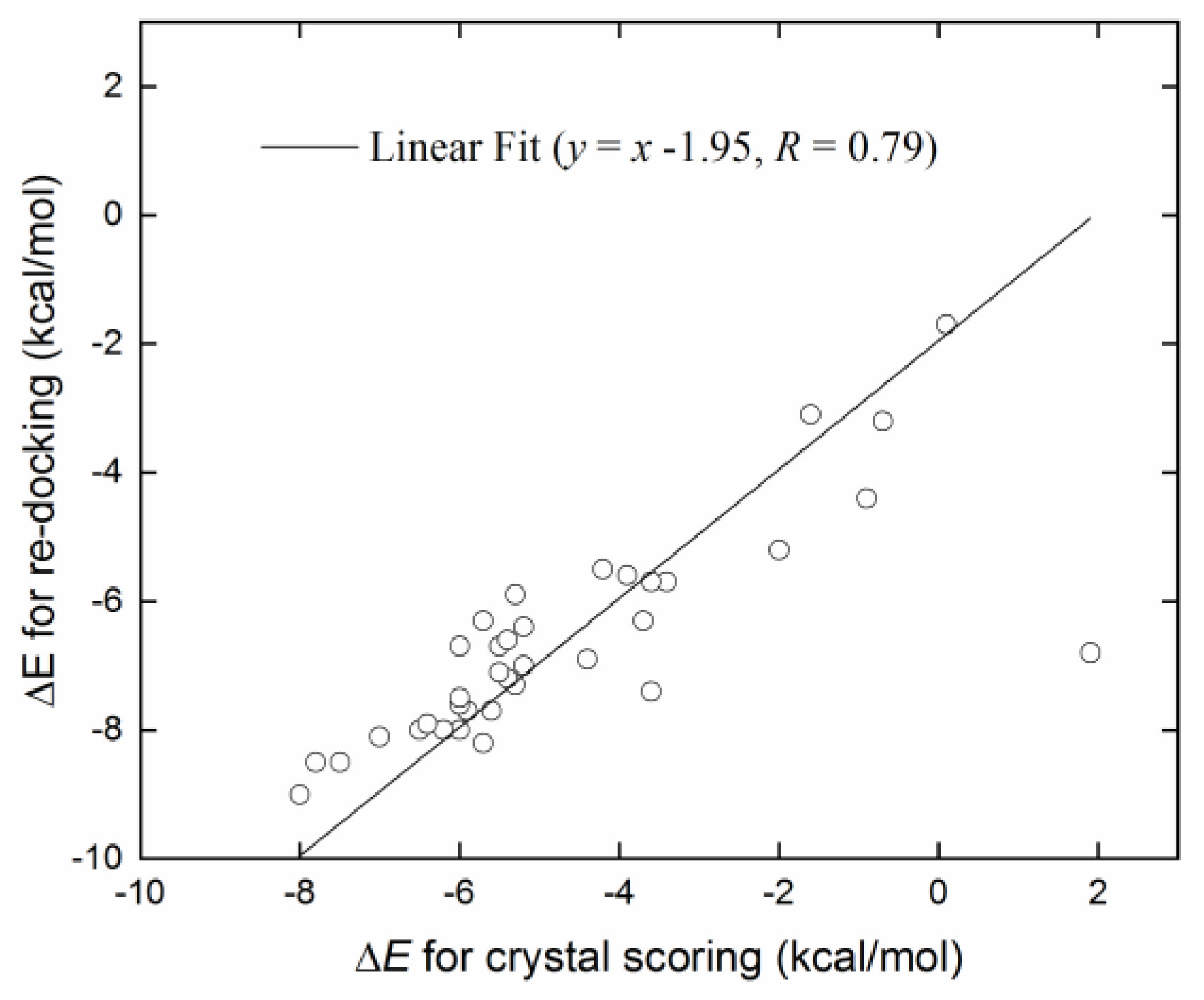
2.1. Evaluation of Crystal Receptor-Ligand Complexes
2.2. Virtual Screening of World-Approved Drugs against hCA I
2.3. Toxicity Evaluation
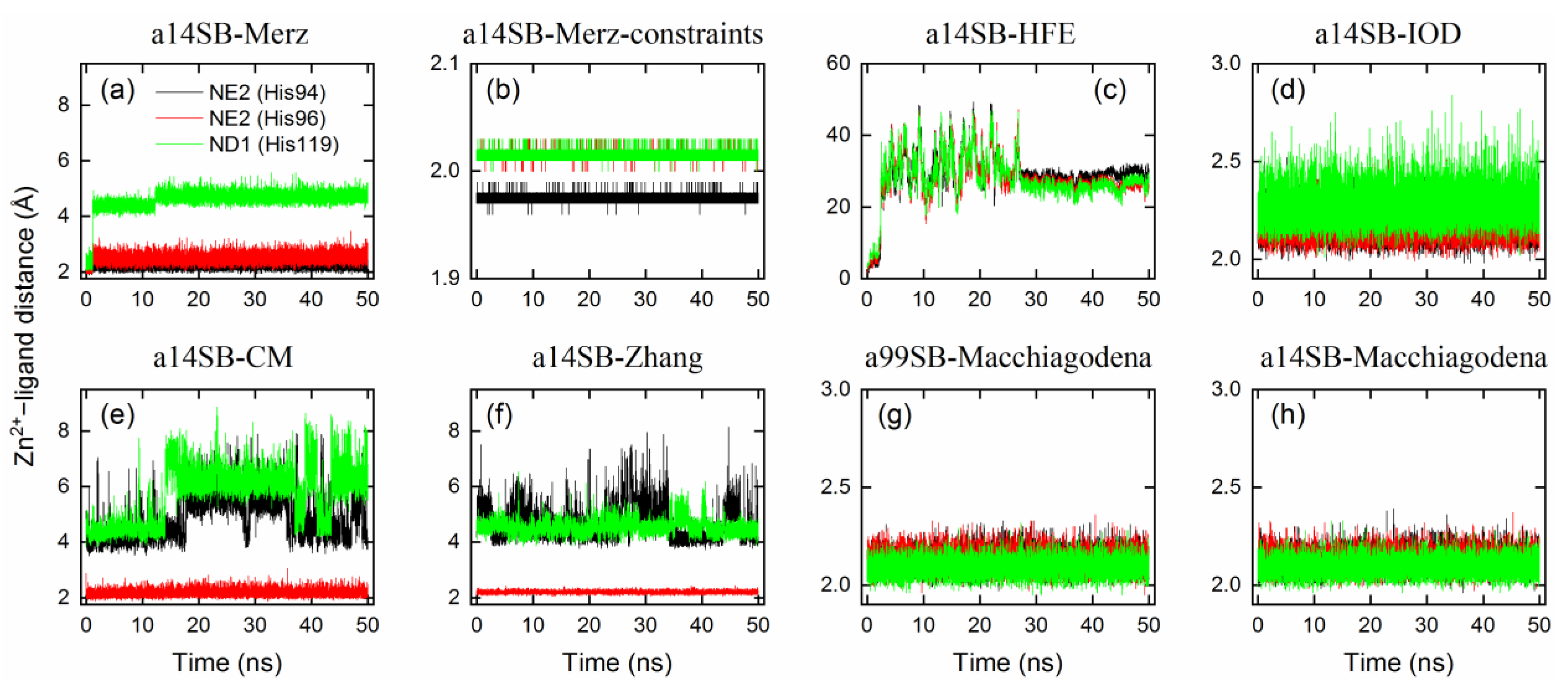
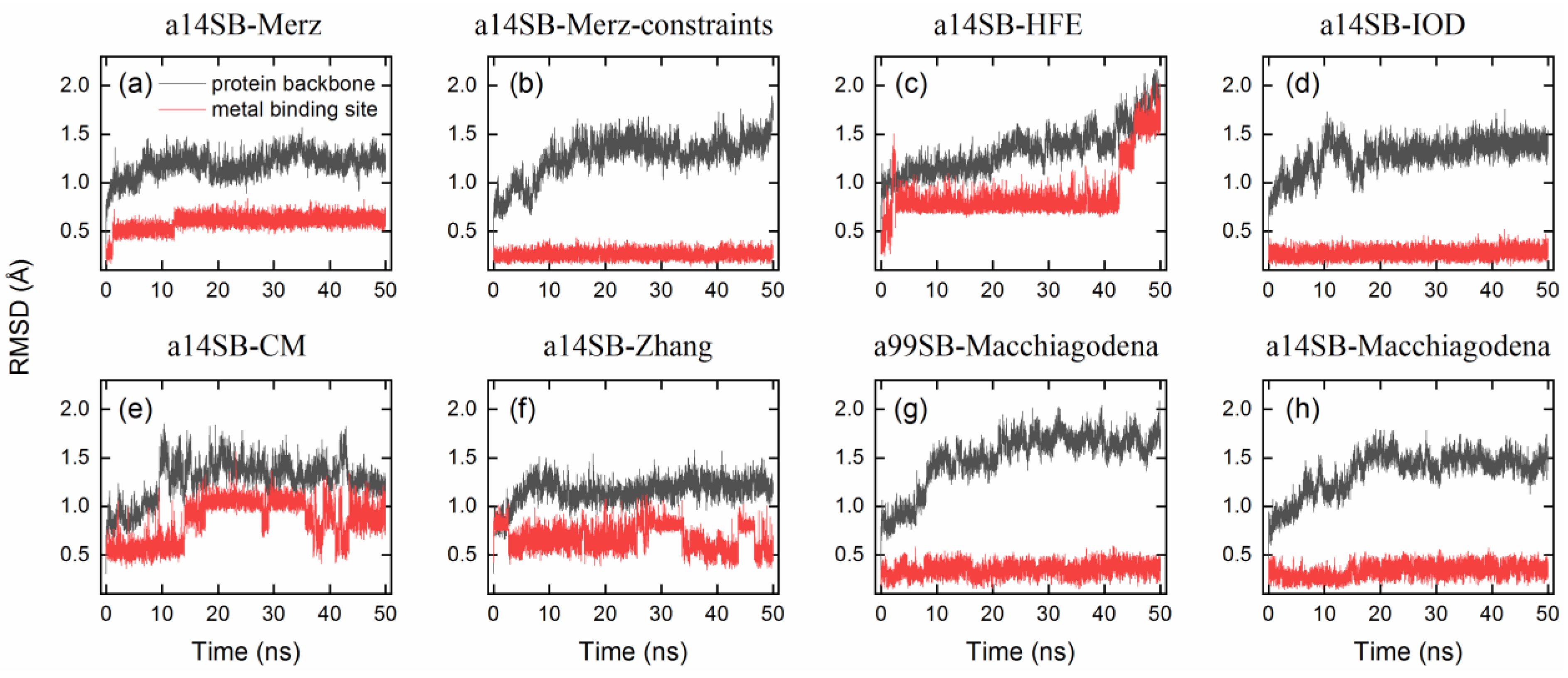
2.4. Benchmark of Zn2+ Ion Models
2.5. MD Simulation of hCA I–Inhibitor Complexes and Binding Energy Calculations
2.6. Identification of Key Residues for Receptor–Inhibitor Interactions
2.7. Selectivity against hCA Isoforms
3. Materials and Methods
3.1. Docking Protocol
3.1.1. Receptor Preparation
3.1.2. Ligand Preparation
3.1.3. Docking Calculation
3.2. Toxicity Prediction
3.3. Simulation Protocol
3.3.1. Ligand-Free Systems
3.3.2. Receptor–Ligand Complexes
3.4. MM-PBSA Analysis
3.5. Inhibition against hCA Isoforms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hassan, I.; Shajee, B.; Waheed, A.; Ahmad, F.; Sly, W.S. Structure, function and applications of carbonic anhydrase isozymes. Bioorg. Med. Chem. 2012, 21, 1570–1582. [Google Scholar] [CrossRef]
- Clare, B.W.; Supuran, C.T. A perspective on quantitative structure–activity relationships and carbonic anhydrase inhibitors. Expert Opin. Drug Metab. Toxicol. 2006, 2, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Moi, D.; Vittorio, S.; Angeli, A.; Balboni, G.; Supuran, C.T.; Onnis, V. Investigation on Hydrazonobenzenesulfonamides as Human Carbonic Anhydrase I, II, IX and XII Inhibitors. Molecules 2022, 28, 91. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 759–772. [Google Scholar] [CrossRef]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Langella, E.; Esposito, D.; Monti, S.M.; Supuran, C.T.; De Simone, G.; Alterio, V. A Combined in Silico and Structural Study Opens New Perspectives on Aliphatic Sulfonamides, a Still Poorly Investigated Class of CA Inhibitors. Biology 2023, 12, 281. [Google Scholar] [CrossRef]
- Pastorekova, S.; Ratcliffe, P.J.; Pastorek, J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. BJU Int. 2008, 101, 8–15. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T. Glaucoma and the Applications of Carbonic Anhydrase Inhibitors. Subcell Biochem. 2013, 75, 349–359. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Fernández-Fernández, C.; Sánchez-Bello, R.; Donapetry-García, C.; Martínez-Rodríguez, J. The role of carbonic anhydrase in the pathogenesis of vascular calcification in humans. Atherosclerosis 2015, 241, 183–191. [Google Scholar] [CrossRef]
- Lobo, M.E.D.V.; Weir, N.; Hardowar, L.; Al Ojaimi, Y.; Madden, R.; Gibson, A.; Bestall, S.M.; Hirashima, M.; Schaffer, C.B.; Donaldson, L.F.; et al. Hypoxia-induced carbonic anhydrase mediated dorsal horn neuron activation and induction of neuropathic pain. Pain 2022, 163, 2264–2279. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Scozzafava, A.; Supuran, C.T. Which carbonic anhydrases are targeted by the antiepileptic sulfonamides and sulfamates? Chem. Biol. Drug Des. 2009, 74, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wei, Y.; Wang, J.; Pi, L.; Huang, J.; Wang, P. Carbonic Anhydrases III and IV Autoantibodies in Rheumatoid Arthritis, Systemic Lupus Erythematosus, Diabetes, Hypertensive Renal Disease, and Heart Failure. Clin. Dev. Immunol. 2012, 2012, 354594. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev. Neurother. 2016, 16, 961–968. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.M.; Supuran, C.T.; De Simone, G. Carbonic Anhydrase IX as a Target for Designing Novel Anticancer Drugs. Curr. Med. Chem. 2012, 19, 821–830. [Google Scholar] [CrossRef]
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef]
- Bayram, E.; Senturk, M.; Kufrevioglu, O.I.; Supuran, C.T. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg. Med. Chem. 2008, 16, 9101–9105. [Google Scholar] [CrossRef]
- Zakšauskas, A.; Čapkauskaitė, E.; Paketurytė-Latvė, V.; Smirnov, A.; Leitans, J.; Kazaks, A.; Dvinskis, E.; Stančaitis, L.; Mickevičiūtė, A.; Jachno, J.; et al. Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX. Int. J. Mol. Sci. 2021, 23, 130. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Abdelrahman, M.A.; Nocentini, A.; Bua, S.; Abdel-Aziz, H.A.; Supuran, C.T.; Abou-Seri, S.M.; Eldehna, W.M. Insights into the effect of elaborating coumarin-based aryl enaminones with sulfonamide or carboxylic acid functionality on carbonic anhydrase inhibitory potency and selectivity. Bioorg. Chem. 2022, 126, 105888. [Google Scholar] [CrossRef]
- Yıldırım, A.; Atmaca, U.; Keskin, A.; Topal, M.; Çelik, M.; Gülçin, I.; Supuran, C.T. N-Acylsulfonamides strongly inhibit human carbonic anhydrase isoenzymes I and II. Bioorg. Med. Chem. 2015, 23, 2598–2605. [Google Scholar] [CrossRef]
- Akıncıoğlu, A.; Akbaba, Y.; Göçer, H.; Göksu, S.; Gülçin, I.; Supuran, C.T. Novel sulfamides as potential carbonic anhydrase isoenzymes inhibitors. Bioorg. Med. Chem. 2013, 21, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef]
- Mincione, F.; Nocentini, A.; Supuran, C.T. Advances in the discovery of novel agents for the treatment of glaucoma. Expert Opin. Drug Discov. 2021, 16, 1209–1225. [Google Scholar] [CrossRef]
- Arechederra, R.L.; Waheed, A.; Sly, W.S.; Supuran, C.T.; Minteer, S.D. Effect of sulfonamides as carbonic anhydrase VA and VB inhibitors on mitochondrial metabolic energy conversion. Bioorg. Med. Chem. 2012, 21, 1544–1548. [Google Scholar] [CrossRef]
- Supuran, C.T. Anti-obesity carbonic anhydrase inhibitors: Challenges and opportunities. J. Enzym. Inhib. Med. Chem. 2022, 37, 2478–2488. [Google Scholar] [CrossRef]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Keller, U.A.D.; Leung, S.; Huntsman, D.; et al. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX Inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditte, P.; Dequiedt, F.; Svastova, E.; Hulikova, A.; Ohradanova-Repic, A.; Zatovicova, M.; Csaderova, L.; Kopacek, J.; Supuran, C.T.; Pastorekova, S.; et al. Phosphorylation of Carbonic Anhydrase IX Controls Its Ability to Mediate Extracellular Acidification in Hypoxic Tumors. Cancer Res. 2011, 71, 7558–7567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, M.A.; Shestakov, A.N.; Zibinsky, M.; Krasavin, M.; Supuran, C.T.; Kalinin, S.; Tanç, M. Synthesis, structure and properties of N -aminosaccharin—A selective inhibitor of human carbonic anhydrase I. Tetrahedron Lett. 2016, 58, 172–174. [Google Scholar] [CrossRef]
- Sechi, M.; Innocenti, A.; Pala, N.; Rogolino, D.; Carcelli, M.; Scozzafava, A.; Supuran, C.T. Inhibition of alpha-class cytosolic human carbonic anhydrases I, II, IX and XII, and beta-class fungal enzymes by carboxylic acids and their derivatives: New isoform-I selective nanomolar inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5801–5806. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.-B.; Clermont, A.; Rook, S.; Fonda, S.J.; Srinivasan, V.J.; Wojtkowski, M.; Fujimoto, J.G.; Avery, R.L.; Arrigg, P.G.; Bursell, S.-E.; et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med. 2007, 13, 181–188. [Google Scholar] [CrossRef]
- Ahamad, S.; Hassan, M.I.; Dwivedi, N. Designing of phenol-based beta-carbonic anhydrase1 inhibitors through QSAR, molecular docking, and MD simulation approach. 3 Biotech 2018, 8, 256. [Google Scholar] [CrossRef]
- Berrino, E.; Bua, S.; Mori, M.; Botta, M.; Murthy, V.S.; Vijayakumar, V.; Tamboli, Y.; Bartolucci, G.; Mugelli, A.; Cerbai, E.; et al. Novel Sulfamide-Containing Compounds as Selective Carbonic Anhydrase I Inhibitors. Molecules 2017, 22, 1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gündüzalp, A.B.; Parlakgümüş, G.; Uzun, D.; Özmen, Ü.Ö.; Özbek, N.; Sarı, M.; Tunç, T. Carbonic anhydrase inhibitors: Synthesis, characterization and inhibition activities of furan sulfonylhydrazones against carbonic anhydrase I (hCA I). J. Mol. Struct. 2016, 1105, 332–340. [Google Scholar] [CrossRef]
- Türkoğlu, E.A.; Şentürk, M.; Supuran, C.T.; Ekinci, D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J. Enzym. Inhib. Med. Chem. 2017, 32, 74–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreyre, H.; Coustard, J.-M.; Carré, G.; Vandebrouck, C.; Bescond, J.; Ouédraogo, M.; Marrot, J.; Vullo, D.; Supuran, C.T.; Thibaudeau, S. Natural product hybrid and its superacid synthesized analogues: Dodoneine and its derivatives show selective inhibition of carbonic anhydrase isoforms I, III, XIII and XIV. Bioorg. Med. Chem. 2013, 21, 3790–3794. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.K.; Sahoo, S.K.; Pichikala, J.N.; Suresh, P. Carbonic anhydrase I and II inhibition with natural products: Caffeine and piperine. J. Enzym. Inhib. Med. Chem. 2011, 27, 97–100. [Google Scholar] [CrossRef]
- Artunç, T.; Çetinkaya, Y.; Göçer, H.; Gülçin, I.; Menzek, A.; Şahin, E.; Supuran, C.T. Synthesis of 4-[2-(3,4-dimethoxybenzyl)cyclopentyl]-1,2-dimethoxybenzene Derivatives and Evaluations of Their Carbonic Anhydrase Isoenzymes Inhibitory Effects. Chem. Biol. Drug Des. 2015, 87, 594–607. [Google Scholar] [CrossRef]
- Casini, A.; Antel, J.; Abbate, F.; Scozzafava, A.; David, S.; Waldeck, H.; Schäfer, S.; Supuran, C.T. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg. Med. Chem. Lett. 2003, 13, 841–845. [Google Scholar] [CrossRef]
- Wagner, J.; Avvaru, B.S.; Robbins, A.H.; Scozzafava, A.; Supuran, C.T.; McKenna, R. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: Solution and crystallographic investigations. Bioorg. Med. Chem. 2010, 18, 4873–4878. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic Anhydrase Inhibition/Activation: Trip of a Scientist Around the World in the Search of Novel Chemotypes and Drug Targets. Curr. Pharm. Des. 2010, 16, 3233–3245. [Google Scholar] [CrossRef]
- Carta, F.; Vullo, D.; Maresca, A.; Scozzafava, A.; Supuran, C.T. Mono-/dihydroxybenzoic acid esters and phenol pyridinium derivatives as inhibitors of the mammalian carbonic anhydrase isoforms I, II, VII, IX, XII and XIV. Bioorg. Med. Chem. 2013, 21, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, F.; Maresca, A.; Scozzafava, A.; Supuran, C.T. 5- and 6-Membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2011, 22, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Vullo, D.; Maresca, A.; Scozzafava, A.; Supuran, C.T. New chemotypes acting as isozyme-selective carbonic anhydrase inhibitors with low affinity for the offtarget cytosolic isoform II. Bioorg. Med. Chem. Lett. 2012, 22, 2182–2185. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Alterio, V.; Supuran, C.T. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2013, 8, 793–810. [Google Scholar] [CrossRef]
- Temperini, C.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L- and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: Stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J. Med. Chem. 2006, 49, 3019–3027. [Google Scholar]
- Durdagi, S.; Şentürk, M.; Ekinci, D.; Balaydın, H.T.; Göksu, S.; Küfrevioğlu, I.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg. Med. Chem. 2011, 19, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, C.; McNamee, A.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Merz, K.M. Carbon dioxide binding to human carbonic anhydrase II. J. Am. Chem. Soc. 1991, 113, 406–411. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. 2013, 10, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jiang, Y.; Peng, J.; Zhang, H. Rational Design of Nonbonded Point Charge Models for Divalent Metal Cations with Lennard-Jones 12-6 Potential. J. Chem. Inf. Model. 2021, 61, 4031–4044. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgrading and Validation of the AMBER Force Field for Histidine and Cysteine Zinc(II)-Binding Residues in Sites with Four Protein Ligands. J. Chem. Inf. Model. 2019, 59, 3803–3816. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgraded AMBER Force Field for Zinc-Binding Residues and Ligands for Predicting Structural Properties and Binding Affinities in Zinc-Proteins. ACS Omega 2020, 5, 15301–15310. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Melse, O.; Antes, I.; Kaila, V.R.I.; Zacharias, M. Benchmarking biomolecular force field-based Zn2+ for mono- and bimetallic ligand binding sites. J. Comput. Chem. 2023, 44, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Zhang, Y.; Jiang, W.; Zhang, H. Virtual Screening of FDA-Approved Drugs for Enhanced Binding with Mitochondrial Aldehyde Dehydrogenase. Molecules 2022, 27, 8773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiu, Y.; Zhang, H. Computational Investigation of Structural Basis for Enhanced Binding of Isoflavone Analogues with Mitochondrial Aldehyde Dehydrogenase. ACS Omega 2022, 7, 8115–8127. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Valdés-Tresanco, M.S.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Res Name | PDB ID | Ligand Name | Molecular Structure | Crystal Complex | Docking Predictions | ||||
|---|---|---|---|---|---|---|---|---|---|
| ∆E | d | ∆E | d | RMSD | Prob | ||||
| 949 | 5GMM | Polmacoxib | 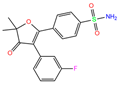 | −8.0 | 1.95 | −9.0 | 3.41 | 3.95 | 1 |
| TOR | 3LXE | Topiramate |  | −7.5 | 2.00 | −8.5 | 2.73 | 4.35 | 1 |
| GZE | 6I0J | GZE |  | −7.8 | 2.00 | −8.5 | 2.55 | 0.73 | 0.7 |
| V14 | 5E2M | V14 | 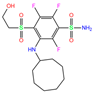 | −5.7 | 1.95 | −8.2 | 2.91 | 1.36 | 1 |
| BZW | 6EVR | BZW | 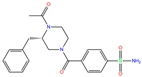 | −7.0 | 1.94 | −8.1 | 4.96 | 1.34 | 1 |
| IWE | 7ZL5 | Diart | 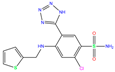 | −6.0 | 1.98 | −8.0 | 2.86 | 1.12 | 1 |
| N19 | 6EX1 | N19 |  | −6.2 | 1.92 | −8.0 | 4.88 | 1.95 | 1 |
| D3B | 6FAF | D3B |  | −6.5 | 1.96 | −8.0 | 4.74 | 1.23 | 0.3 |
| 3UG | 4WUQ | 3UG | 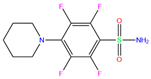 | −6.4 | 1.84 | −7.9 | 2.92 | 1.25 | 1 |
| CJK | 6F3B | CJK |  | −5.6 | 1.93 | −7.7 | 4.58 | 2.06 | 1 |
| O5N | 6XZY | O5N | 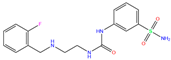 | −5.9 | 1.84 | −7.7 | 3.15 | 3.33 | 0.8 |
| EON | 6FAG | EON | 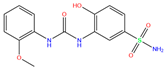 | −6.0 | 1.90 | −7.6 | 2.47 | 2.20 | 0.6 |
| O5H | 6XZX | O5H | 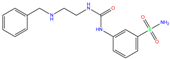 | −6.0 | 1.90 | −7.5 | 2.96 | 2.50 | 0.4 |
| 84Z | 7Q0D | 84Z |  | −3.6 | 1.91 | −7.4 | 3.96 | 2.43 | 0.9 |
| O4Z | 6XZE | O4Z |  | −5.3 | 1.95 | −7.3 | 4.11 | 4.09 | 1 |
| O5K | 6XZS | O5K |  | −5.4 | 1.89 | −7.2 | 4.24 | 4.73 | 0.7 |
| 3TV | 4WR7 | 3TV |  | −5.5 | 1.84 | −7.1 | 3.12 | 1.34 | 1 |
| O55 | 6XZO | O55 | 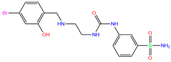 | −5.2 | 1.93 | −7.0 | 3.07 | 7.43 | 0.9 |
| 7TI | 7PLF | Clorsulon | 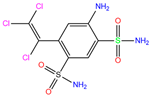 | −4.4 | 1.94 | −6.9 | 2.85 | 3.23 | 1 |
| FLB | 3W6I | FLB |  | 1.9 | 5.06 | −6.8 | 7.31 | 3.19 | 1 |
| O5T | 6Y00 | O5T |  | −6.0 | 1.92 | −6.7 | 4.99 | 1.57 | 0.1 |
| M25 | 2NMX | M25 | 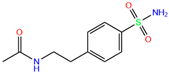 | −5.5 | 2.19 | −6.7 | 2.31 | 2.72 | 1 |
| M29 | 2NN7 | M29 |  | −5.4 | 2.11 | −6.6 | 2.36 | 3.22 | 1 |
| M28 | 2NN1 | M28 | 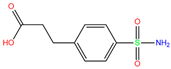 | −5.2 | 2.21 | −6.4 | 2.61 | 2.67 | 1 |
| AZM | 3W6H | Acetazolamide | 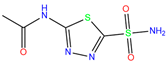 | −3.7 | 2.17 | −6.3 | 2.29 | 2.57 | 0.1 |
| MZM | 1BZM | Methazola-mide | 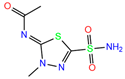 | −5.7 | 1.99 | −6.3 | 2.88 | 1.32 | 0.6 |
| AZM | 1AZM | Acetazola-mide | 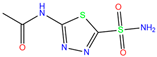 | −5.3 | 2.01 | −5.9 | 3.68 | 2.22 | 1 |
| 3UF | 4WUP | 3UF | 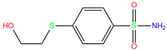 | −3.4 | 1.74 | −5.7 | 16.21 | 15.72 | 1 |
| FO9 | 6G3V | Famotidine |  | −3.6 | 1.95 | −5.7 | 16.82 | 15.52 | 1 |
| AAS | 1CZM | 3-Amabs |  | −3.9 | 1.86 | −5.6 | 2.42 | 2.24 | 1 |
| GZH | 6I0L | GZH |  | −4.2 | 1.92 | −5.5 | 3.96 | 1.84 | 1 |
| HIS | 2FW4 | d-histidine | 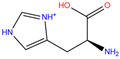 | −2.0 | 29.79 | −5.2 | 16.13 | 16.13 | 1 |
| PPF | 2IT4 | Foscarnet | 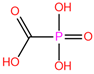 | −0.9 | 1.73 | −4.4 | 13.08 | 15.59 | 1 |
| EDO | 1JV0 | Ethylene glycol |  | −0.7 | 1.82 | −3.2 | 4.59 | 6.04 | 0.8 |
| BCT | 1HCB | Carbonate |  | −1.6 | 1.78 | −3.1 | 16.17 | 12.89 | 1 |
| EDO | 1J9W | Ethylene glycol |  | 0.1 | 1.75 | −1.7 | 14.26 | 13.57 | 1 |
| ZINC ID | Name | Molecular Structure | q | ∆Edock | Toxicity | ||||
|---|---|---|---|---|---|---|---|---|---|
| Dili | Carcino | Immuno | Mutagen | Cyto | |||||
| ZINC000011681563 | Netupitant |  | 0 | −9.9 | N(66) | N(71) | N(78) | N(73) | N(64) |
| ZINC000011681563 | Netupitant | 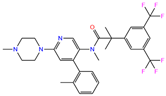 | 1 | −9.9 | N(66) | N(71) | N(78) | N(73) | N(64) |
| ZINC000000601301 | Bhft |  | 0 | −9.8 | N82) | N(71) | N(89) | N(84) | N73) |
| ZINC000205224698 | Pronetupitant | 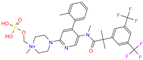 | 1 | −9.8 | N(66) | N(58) | N(53) | N(55) | N(61) |
| ZINC000027990463 | Lomitapide | 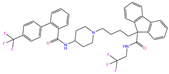 | 1 | −9.7 | N(78) | N(61) | N(70) | N(58) | N(77) |
| ZINC000021981256 | 8-hydroxymirtazapine | 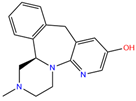 | 1 | −9.6 | N(83) | N(66) | N(96) | N(61) | N(59) |
| ZINC000003816514 | Rolapitant |  | 0 | −9.6 | N(83) | N(66) | N(93) | N(67) | N(75) |
| ZINC000003816514 | Rolapitant |  | 1 | −9.6 | N(83) | N(66) | N(93) | N(67) | N(75) |
| ZINC000022034381 | Lidoflazine | 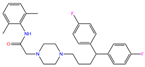 | 0 | −9.5 | N(79) | N(70) | N(97) | N(78) | N(67) |
| ZINC000031417974 | Ketoprofen glucuronide |  | −1 | −9.3 | N(71) | N(68) | N(85) | N(85) | N(82) |
| ZINC000002570882 | 4-hydroxyalprazolam | 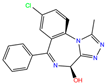 | 0 | −9.2 | N(52) | N(67) | N(97) | N(74) | N(70) |
| ZINC000000607726 | Bemetizide | 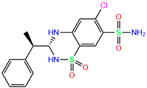 | 0 | −9.2 | N(89) | N(76) | N(88) | N(92) | N(81) |
| ZINC000022034381 | Lidoflazine |  | 1 | −9.2 | N(79) | N(70) | N(97) | N(78) | N(67) |
| ZINC000100036907 | Cyclothiazide |  | 0 | −9.1 | N(91) | N(74) | N(91) | N(88) | N(73) |
| ZINC000000589683 | Polmacoxib |  | 0 | −9.0 | N(53) | N(62) | N(89) | N(70) | N(67) |
| ZINC0000005843546 | Diart | 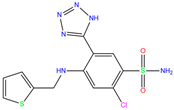 | 0 | −8.0 | N(59) | N(53) | N(95) | N(69) | N(62) |
| ZINC0000263621146 | 3UG |  | 0 | −7.9 | N(66) | N(58) | N(99) | N(81) | N(69) |
| 84Z | 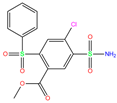 | 0 | −7.4 | N(65) | N(67) | N(99) | N(75) | N(70) | |
| Legend | Force Field | Zn2+ Model | RMSDbackbone | RMSDmetal | Zn2+–Ligand Distance (Å) | CNZn | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter Set | R | ε | Ref | (Å) | (Å) | NE2 (H94) | NE2 (H96) | ND1 (H119) | |||
| a14SB-Merz | ff14SB | Merz | 1.1 | 0.0125 | [50] | 1.25 ± 0.08 | 0.62 ± 0.05 | 2.25 ± 0.14 | 2.54 ± 0.19 | 4.71 ± 0.17 | 4.4 |
| a14SB-Merz-constraints | ff14SB | Merz (constraints) | 1.1 | 0.0125 | [50] | 1.37 ± 0.12 | 0.26 ± 0.04 | 1.98 ± 0.01 | 2.02 ± 0.01 | 2.02 ± 0.01 | 6.0 |
| a14SB-HFE | ff14SB | HFE set by Li et al. | 1.175 | 0.00071558 | [51] | 1.55 ± 0.19 | 1.05 ± 0.37 | 28.57 ± 1.45 | 26.31 ± 1.25 | 25.69 ± 1.54 | 3.0 |
| a14SB-IOD | ff14SB | IOD set by Li et al. | 1.395 | 0.014917 | [51] | 1.38 ± 0.08 | 0.28 ± 0.05 | 2.17 ± 0.07 | 2.18 ± 0.07 | 2.28 ± 0.09 | 6.0 |
| a14SB-CM | ff14SB | CM set by Li et al. | 1.271 | 0.00330286 | [51] | 1.32 ± 0.12 | 0.91 ± 0.16 | 5.00 ± 0.80 | 2.20 ± 0.13 | 6.10 ± 0.83 | 6.0 |
| a14SB-Zhang | ff14SB | Zhang et al. | 0.5152 | 295.5289 | [52] | 1.21 ± 0.08 | 0.65 ± 0.14 | 4.57 ± 0.59 | 2.20 ± 0.05 | 4.55 ± 0.28 | 7.0 |
| a99SB-Macchiagodena | ff99SB-ILDN | Macchiagodena et al. | 1.4561 | 0.0125 | [53,54] | 1.69 ± 0.10 | 0.36 ± 0.07 | 2.12 ± 0.05 | 2.12 ± 0.05 | 2.09 ± 0.04 | 6.0 |
| a14SB-Macchiagodena | ff14SB | Macchiagodena et al. | 1.4561 | 0.0125 | [53,54] | 1.46 ± 0.09 | 0.36 ± 0.06 | 2.12 ± 0.05 | 2.12 ± 0.05 | 2.09 ± 0.04 | 6.0 |
| crystal-7q0d | 1.97 | 2.02 | 2.02 | 4.0 | |||||||
| Ligand Name | q | dML | Ligand Atom | dbound | ||
|---|---|---|---|---|---|---|
| Initial | MD | Initial | MD | |||
| Netupitant | 0 | 1.04 | 1.59 ± 0.11 | F6 | 2.95 | 12.97 ± 2.24 |
| Netupitant | 1 | 1.14 | 2.93 ± 0.56 | F6 | 3.14 | 27.7 ± 5.63 |
| Bhft | 0 | 0.9 | 0.92 ± 0.03 | O1 | 2.53 | 2.33 ± 0.15 |
| N1 | 4.73 | 2.31 ± 0.12 | ||||
| Pronetupitant | 0 | 1.12 | 2.09 ± 0.34 | F6 | 2.83 | 17.81 ± 5.85 |
| Lomitapide | 1 | 1.15 | 1.91 ± 0.38 | F2 | 2.82 | 16.57 ± 4.75 |
| 8-hydroxymirtazapine | 1 | 0.92 | 2.58 ± 0.71 | O1 | 2.55 | 27.58 ± 6.54 |
| Rolapitant | 0 | 0.95 | 1.61 ± 0.11 | F6 | 3.47 | 11.82 ± 1.98 |
| Rolapitant | 1 | 0.94 | 1.49 ± 0.09 | F5 | 2.67 | 11.01 ± 1.41 |
| Lidoflazine | 0 | 0.96 | 1.42 ± 0.06 | F1 | 2.88 | 4.37 ± 0.24 |
| Ketoprofen glucuronide | −1 | 0.99 | 1.08 ± 0.02 | O4 | 3.22 | 2.08 ± 0.06 |
| 4-hydroxyalprazolam | 0 | 0.85 | 1.95 ± 0.04 | N1 | 2.92 | 14.79 ± 0.63 |
| Bemetizide | 0 | 0.81 | 0.85 ± 0.03 | N2 | 2.30 | 2.31 ± 0.12 |
| Lidoflazine | 1 | 0.96 | 1.34 ± 0.13 | F1 | 2.80 | 4.63 ± 0.30 |
| Cyclothiazide | 0 | 0.85 | 0.81 ± 0.02 | N1 | 2.47 | 4.43 ± 0.24 |
| O2 | 2.60 | 2.44 ± 0.21 | ||||
| Polmacoxib | 0 | 0.89 | 0.91 ± 0.04 | N1 | 1.95 | 4.62 ± 0.19 |
| O3 | 2.88 | 2.33 ± 0.17 | ||||
| Diart | 0 | 0.81 | 1.32 ± 0.15 | N5 | 1.98 | 10.39 ± 1.46 |
| 3UG | 0 | 0.82 | 0.83 ± 0.02 | N1 | 1.84 | 2.30 ± 0.10 |
| 84Z | 0 | 0.84 | 0.86 ± 0.02 | N1 | 1.91 | 4.39 ± 0.22 |
| O3 | 2.99 | 2.29 ± 0.13 | ||||
| Compound | q | ΔEvdW | ΔEelec | ΔEMM | ΔGpolar | ΔGnonpolar | ΔEbind |
|---|---|---|---|---|---|---|---|
| 84Z | 0 | −34.25 ± 1.17 | −43.15 ± 2.00 | −77.4 ± 2.15 | 63.13 ± 1.51 | −3.57 ± 0.03 | −17.84 ± 1.80 |
| Cyclothiazide | 0 | −39.51 ± 1.27 | −38.52 ± 1.85 | −78.03 ± 2.20 | 65.26 ± 2.43 | −3.44 ± 0.03 | −16.21 ± 1.85 |
| 3UG | 0 | −24.81 ± 0.56 | −58.7 ± 0.61 | −83.5 ± 0.55 | 72.45 ± 0.95 | −2.93 ± 0.02 | −13.99 ± 0.40 |
| Bhft | 0 | −33.44 ± 0.70 | −68.73 ± 1.72 | −102.16 ± 0.92 | 96.57 ± 1.57 | −3.73 ± 0.05 | −9.33 ± 2.04 |
| Ketoprofen glucuronide | −1 | −35.30 ± 0.29 | −183.62 ± 2.00 | −218.92 ± 0.78 | 213.98 ± 0.88 | −4.17 ± 0.01 | −9.12 ± 0.55 |
| Polmacoxib | 0 | −26.56 ± 0.45 | −27.71 ± 2.57 | −54.28 ± 1.15 | 54.70 ± 1.67 | −3.40 ± 0.02 | −2.98 ± 0.46 |
| Bemetizide | 0 | −32.23 ± 1.69 | −31.56 ± 3.37 | −63.78 ± 2.21 | 68.27 ± 7.94 | −3.61 ± 0.12 | 0.87 ± 2.80 |
| Name | Identifier | 84Z | Cyclothiazide | 3UG |
|---|---|---|---|---|
| hCA I | 7Q0D | −8.0 ± 0.1 | −9.1 ± 0.1 | −7.9 ± 0.1 |
| hCA II | 1BCD | −8.0 ± 0.1 | −8.3 ± 0.1 | −7.4 ± 0.1 |
| hCA III | 3UYN | −6.8 ± 0.1 | −7.5 ± 0.1 | −6.8 ± 0.1 |
| hCA IV | 5IPZ | −6.9 ± 0.1 | −6.5 ± 0.2 | −6.2 ± 0.1 |
| hCA VA | AF-P35218-F1 | −6.2 ± 0.1 | −6.9 ± 0.1 | −5.8 ± 0.2 |
| hCA VB | AF-Q9Y2D0-F1 | −7.0 ± 0.1 | −7.6 ± 0.2 | −6.7 ± 0.1 |
| hCA VI | 3FE4 | −6.0 ± 0.1 | −7.1 ± 0.1 | −6.1 ± 0.1 |
| hCA VII | 6H37 | −7.2 ± 0.1 | −8.1 ± 0.1 | −7.4 ± 0.1 |
| hCA IX | 6FE1 | −7.5 ± 0.1 | −8.2 ± 0.1 | −7.3 ± 0.1 |
| hCA XII | 1JD0 | −7.1 ± 0.2 | −7.6 ± 0.3 | −6.9 ± 0.1 |
| hCA XIII | 4KNM | −7.8 ± 0.1 | −8.9 ± 0.1 | −7.9 ± 0.1 |
| hCA XIV | 4LU3 | −7.8 ± 0.1 | −8.3 ± 0.1 | −7.3 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, N.; Jiang, W.; Zhang, P.; Ma, L.; Chen, J.; Zhang, H. Repurposing of World-Approved Drugs for Potential Inhibition against Human Carbonic Anhydrase I: A Computational Study. Int. J. Mol. Sci. 2023, 24, 12619. https://doi.org/10.3390/ijms241612619
Zheng N, Jiang W, Zhang P, Ma L, Chen J, Zhang H. Repurposing of World-Approved Drugs for Potential Inhibition against Human Carbonic Anhydrase I: A Computational Study. International Journal of Molecular Sciences. 2023; 24(16):12619. https://doi.org/10.3390/ijms241612619
Chicago/Turabian StyleZheng, Nannan, Wanyun Jiang, Puyu Zhang, Le Ma, Junzhao Chen, and Haiyang Zhang. 2023. "Repurposing of World-Approved Drugs for Potential Inhibition against Human Carbonic Anhydrase I: A Computational Study" International Journal of Molecular Sciences 24, no. 16: 12619. https://doi.org/10.3390/ijms241612619
APA StyleZheng, N., Jiang, W., Zhang, P., Ma, L., Chen, J., & Zhang, H. (2023). Repurposing of World-Approved Drugs for Potential Inhibition against Human Carbonic Anhydrase I: A Computational Study. International Journal of Molecular Sciences, 24(16), 12619. https://doi.org/10.3390/ijms241612619