1. Introduction
Plasmodium falciparum (Pf) is a unicellular parasite responsible for the deadliest form of human malaria. It poses a significant threat to global health, particularly in regions where the disease is endemic [
1]. The function of Pf proteins is regulated by various post-translational modifications, with reversible protein phosphorylation being the most common protein modification observed in the parasite. Protein phosphorylation allows cells to adapt their functions rapidly in response to internal and external changes [
2].
Among the Serine (Ser)/Threonine (Thr) phosphatases, Protein Phosphatase 1 (PP1c) (PF3D7_1414400) plays a crucial role in diverse cellular functions in
Plasmodium and other organisms [
3]. PP1c is a highly conserved enzyme in eukaryotes, and
Plasmodium PP1c (PfPP1c) shares approximately 80% identity with its counterparts in mammals. Its phosphatase activity on phosphopeptides and small substrates is conserved across PP1c homologs in many species [
4].
PP1c functions by associating with various regulatory partners to form holoenzymes, which specifically dephosphorylate a wide range of substrates in different cellular locations. Mammalian cells have 200 identified regulatory subunits that contribute to the specificity, location, and level of phosphatase activity of PP1c [
5,
6]. In Pf and in
Plasmodium berghei (Pb), PP1c has been shown to have numerous potential regulatory partners, with hundreds of interacting proteins identified through yeast two-hybrid (Y2H) and immunoprecipitation experiments combined with mass spectrometry analysis [
7,
8,
9].
Among the PP1c-interacting proteins, three conserved regulators (Inhibitor 2, Inhibitor 3, and LRR1) and a
Plasmodium-specific protein Gametocyte EXported Protein 15 (GEXP15) (PF3D7_1031600) were detected as top interactors [
10,
11]. Biochemical studies have shown a direct interaction between PbGEXP15 and PbPP1c, increasing the phosphatase activity of PP1c in vitro [
7]. Knockout of PbGEXP15 in Pb showed the vital role for the protein during the asexual life cycle and the mosquito stages, where no oocysts and sporozoites were found [
7]. This phenotype could be attributed to a decrease in protein dephosphorylation due to the absence of PP1c control in the PbGEXP15 knockout line. Additionally, the crucial role of PbGEXP15 may be related to its interactome, as it was found to be associated with protein complexes involved in essential biological pathways, such as mRNA splicing and the proteasome pathway [
7].
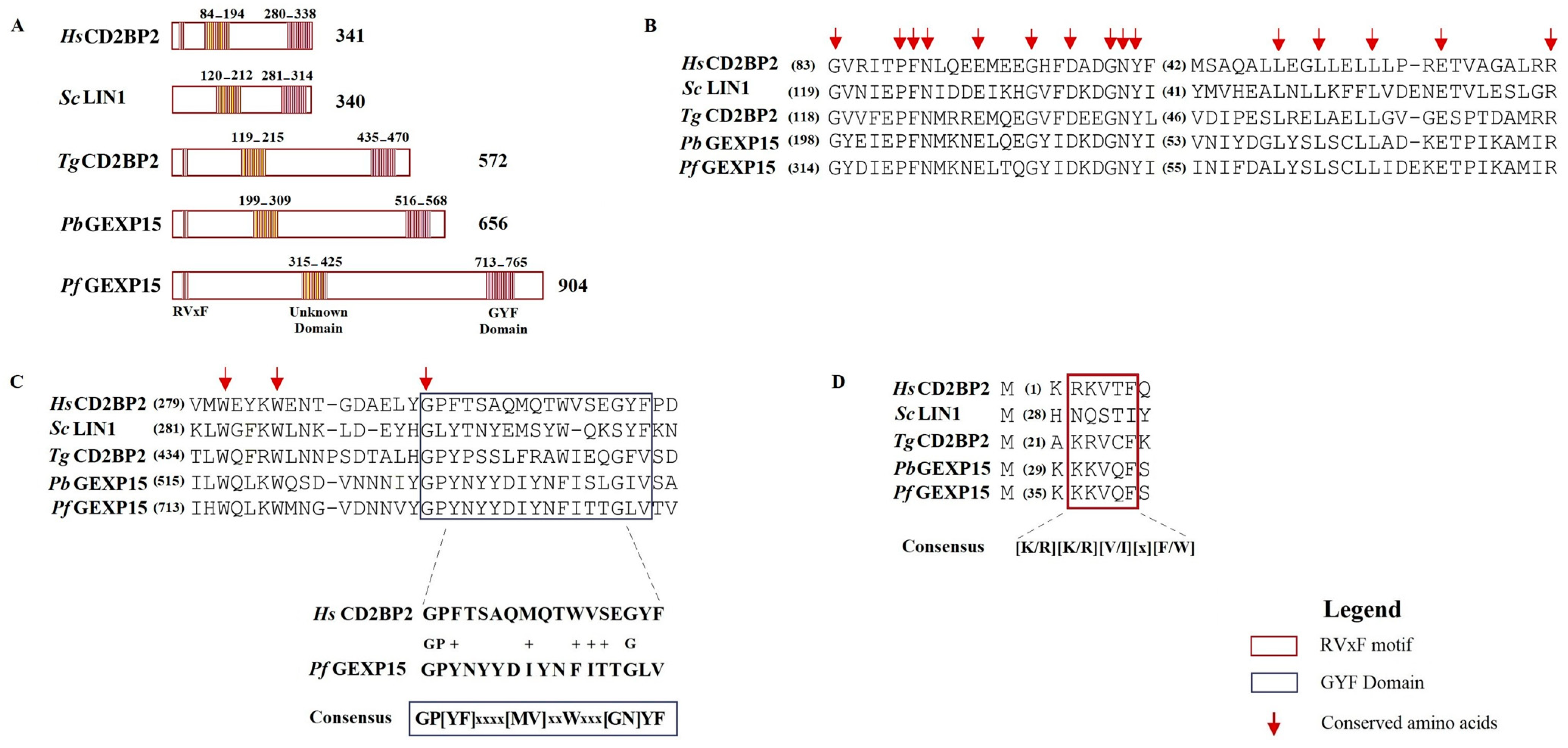
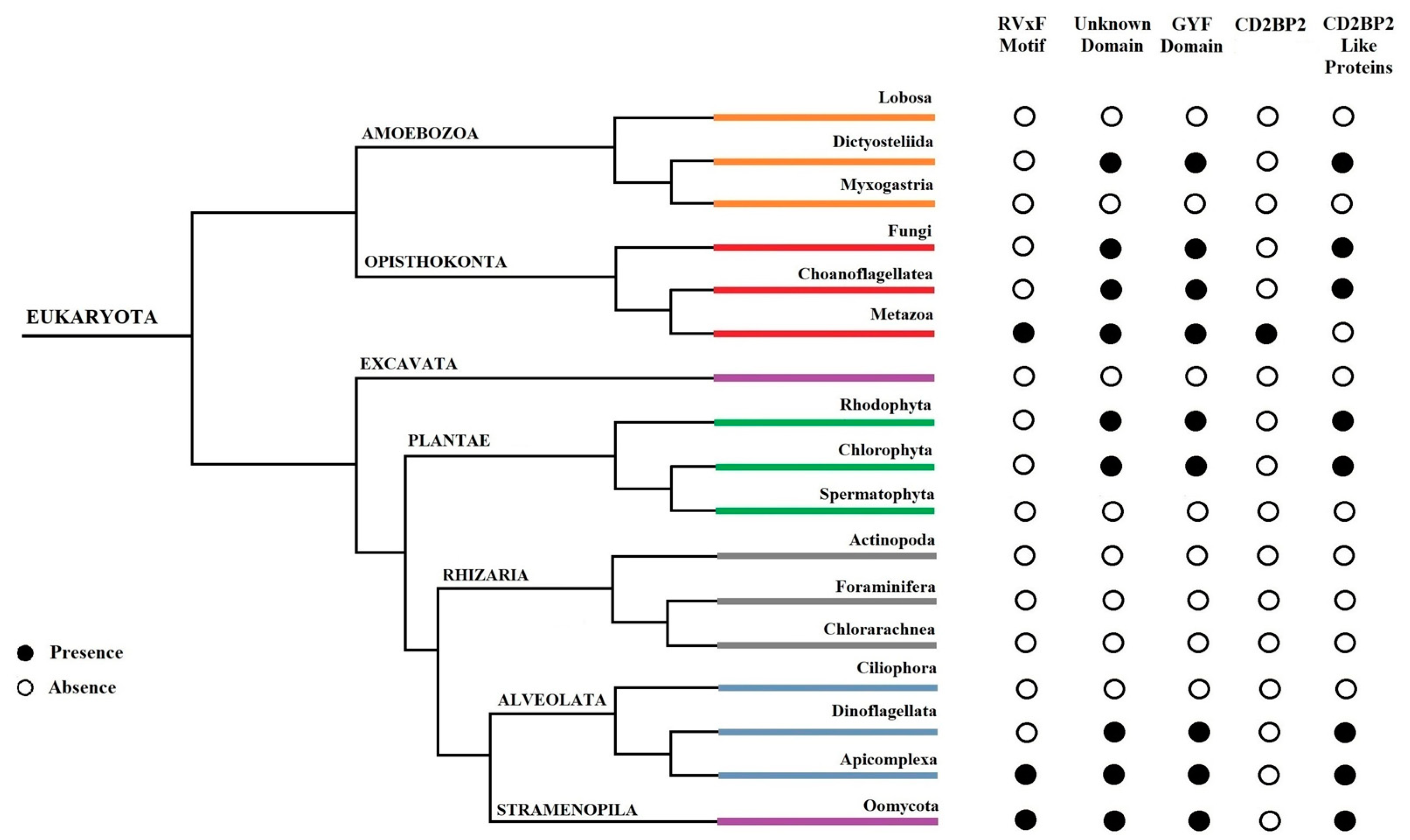
In addition to the RVxF motif located at the N-terminus of PbGEXP15, a GYF domain was identified at its C-terminal [
12]. The GYF domain, characterized by the consensus sequence GP[YF]xxxx[MV]xxWxxx[GN]YF (IPR003169), is known to play a role in protein–protein interactions and is present in numerous proteins in mammals [
13]. The GYF domain was initially described in the CD2 Cytoplasmic Tail-Binding Protein 2 (CD2BP2) expressed in human T cells, where it interacts with the cytoplasmic tail of the CD2 receptor, contributing to T cell activation [
13]. Further studies have indicated that CD2BP2 is also present in the nucleus and may be part of the pre-spliceosomal complex [
14]. Conditional gene targeting in mice revealed the essential role of CD2BP2 in embryonic development [
15]. Based on reciprocal best hits (RBH) analysis, GEXP15 in
Plasmodium is suggested to be an ortholog of human CD2BP2 [
16].
Although studies on proteins functions in Pb, the most tractable of the most rodent malaria models for experimental genetics, can provide valuable insights into fundamental aspects of
Plasmodium biology, there are limits to how much can be extrapolated to Pf [
17]. For instance, targeted gene-by-gene functional studies showed that the gene encoding Schewanella-like phosphatase (shlp1) in Pf was described as likely essential for erythrocyte development by a functional screen analysis [
18]. On the contrary, in Pb, shlp1 is dispensable for the development of blood stage parasites [
19].
In Pf, genome-wide saturation mutagenesis suggested GEXP15 as an essential gene in the intraerythrocytic developmental cycle. However, the specific roles of this protein throughout the lifecycle of Pf are still not fully characterized. In this study, we aimed to investigate the structural and functional characteristics of GEXP15 in Pf. We employed various approaches, including comparative genomics, structural and evolutionary analyses, in vivo studies using an inducible GEXP15 knockdown line to examine cellular localization and function, and protein–protein interaction analyses to explore GEXP15′s interactors and interactome. Through these methods, we uncovered the critical interactome and potential role of GEXP15 in Pf.
3. Discussion
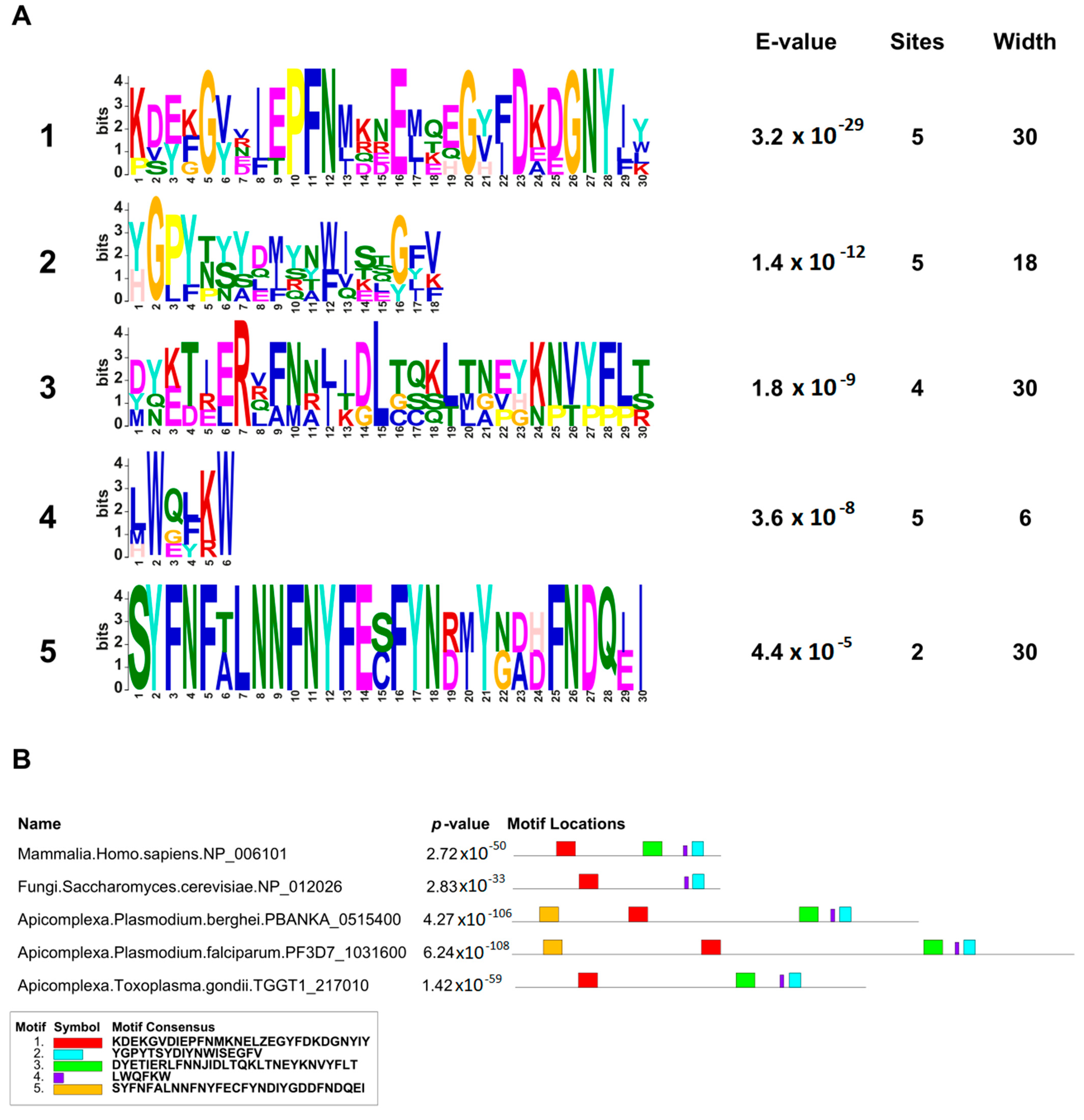
In this work, we provided a better understanding of the structure and evolution of GEXP15 and its homologs in various organisms. A closer examination of these proteins highlighted three regions of particular interest. First, an RVxF motif was detected by manual inspection in the N-terminal region of PfGEXP15, PbGEXP15, TgCD2BP2, and HsCD2BP2. Using the FIMO tool, we confirmed the presence of this motif in various phyla including Apicomplexa, Metazoa, and Nematoda. This motif is known to be implicated in PP1 interaction in eukaryotes and our previous work conducted in
Plasmodium had already established the capacity of PbGEXP15 as well as other regulators to modulate the activity of PP1 [
7,
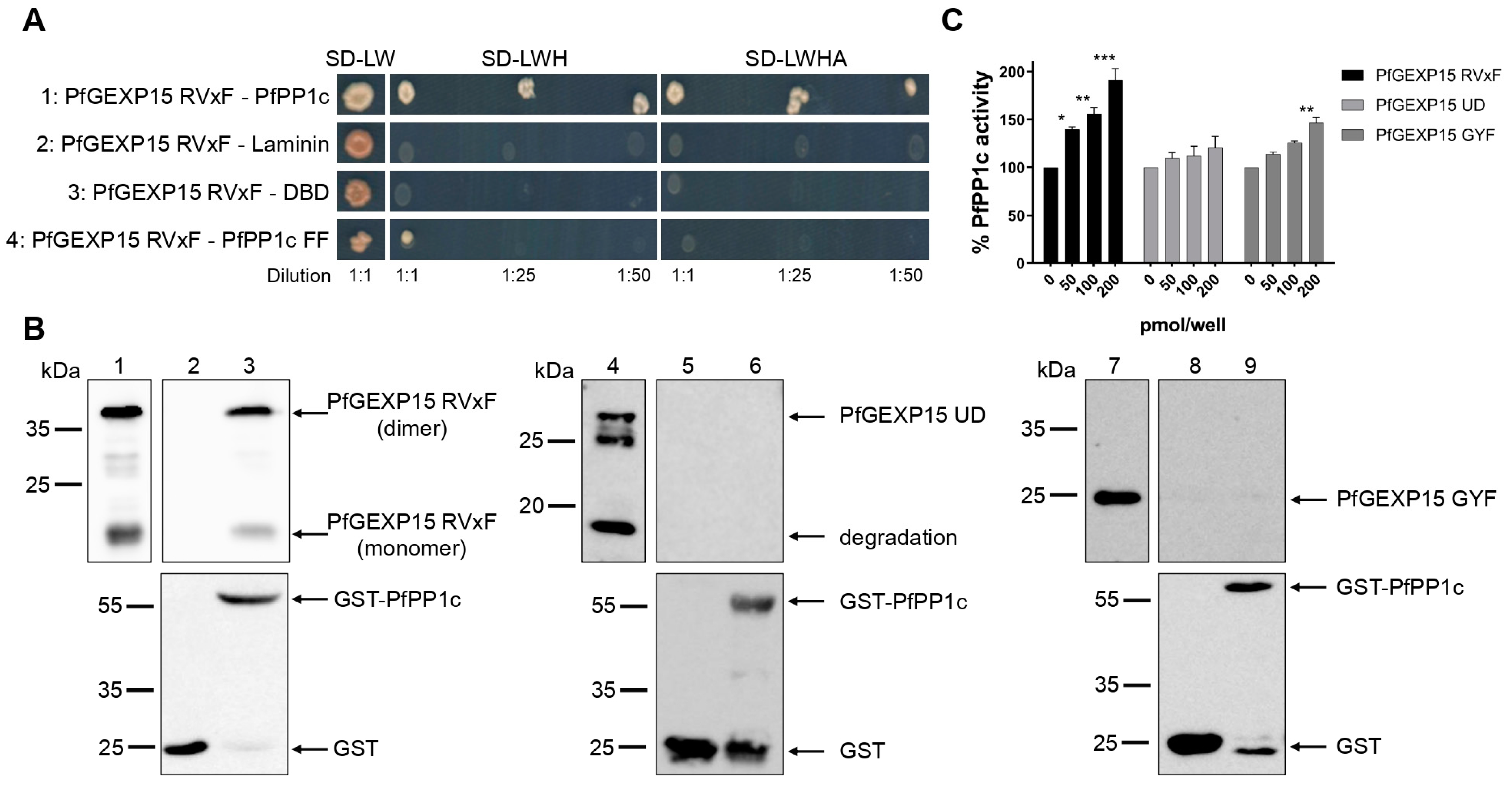
10]. Here, we validated by Y2H and GST pull-down that PfGEXP15 bound to PP1, and that this interaction is RVxF-specific since the PP1 mutant and other GEXP15 regions were not able to interact. Furthermore, we demonstrated the ability of PfGEXP15 to regulate the dephosphorylation activity of PP1 through its N-terminal region containing the RVxF-binding motif. These findings confirmed the preponderant role of the RVxF motif in the interaction and regulation of PP1 by GEXP15.
Second, a conserved domain with an unknown function was identified through the in silico comparative study conducted on the different species as well as with the MEME analysis. Although our pull-down and interactome analyses showed that this domain is unlikely to be involved as a platform for protein interactions, the conservation of critical residues across distant species suggests that this UD region may play a crucial unknown role. From the MS analysis of the pull-down performed with this domain, we found only the exportin 7 (PF3D7_0910100) as a potential binder, which was detected in the nuclear fraction of Pf, suggesting its potential role in PfGEXP15 nuclear trafficking [
31]. In this context, it should be noted that a previous study reported that exportin 5 is required in nuclear export of 60S ribosome subunits in human cells [
33]. Further studies will be necessary for
Plasmodium to elucidate the contribution of this domain to GEXP15 function.
Finally, our in silico study highlighted the presence of a GYF-like domain in GEXP15. The GYF domain is present in a diverse array of proteins, known to interact with proline-rich peptides, including those found in RNA-binding proteins, cytoskeletal proteins, and transcription factors [
14]. Notably, the function of the GYF domain can be modulated by subtle changes in its amino acid sequence, making it a flexible region for regulating protein–protein interactions in a context-dependent manner [
14]. This observation may explain why among the sequences of CB2BP2 and GEXP15 analyzed in this study, only the metazoan proteins had a GYF domain matching the currently described consensus sequence. However, despite the observed differences, the MEME analysis and 3D modeling confirmed some degree of conservation of the GYF-like domains identified in the other species, which may confer adaptation to mediate distinct protein–protein interactions.
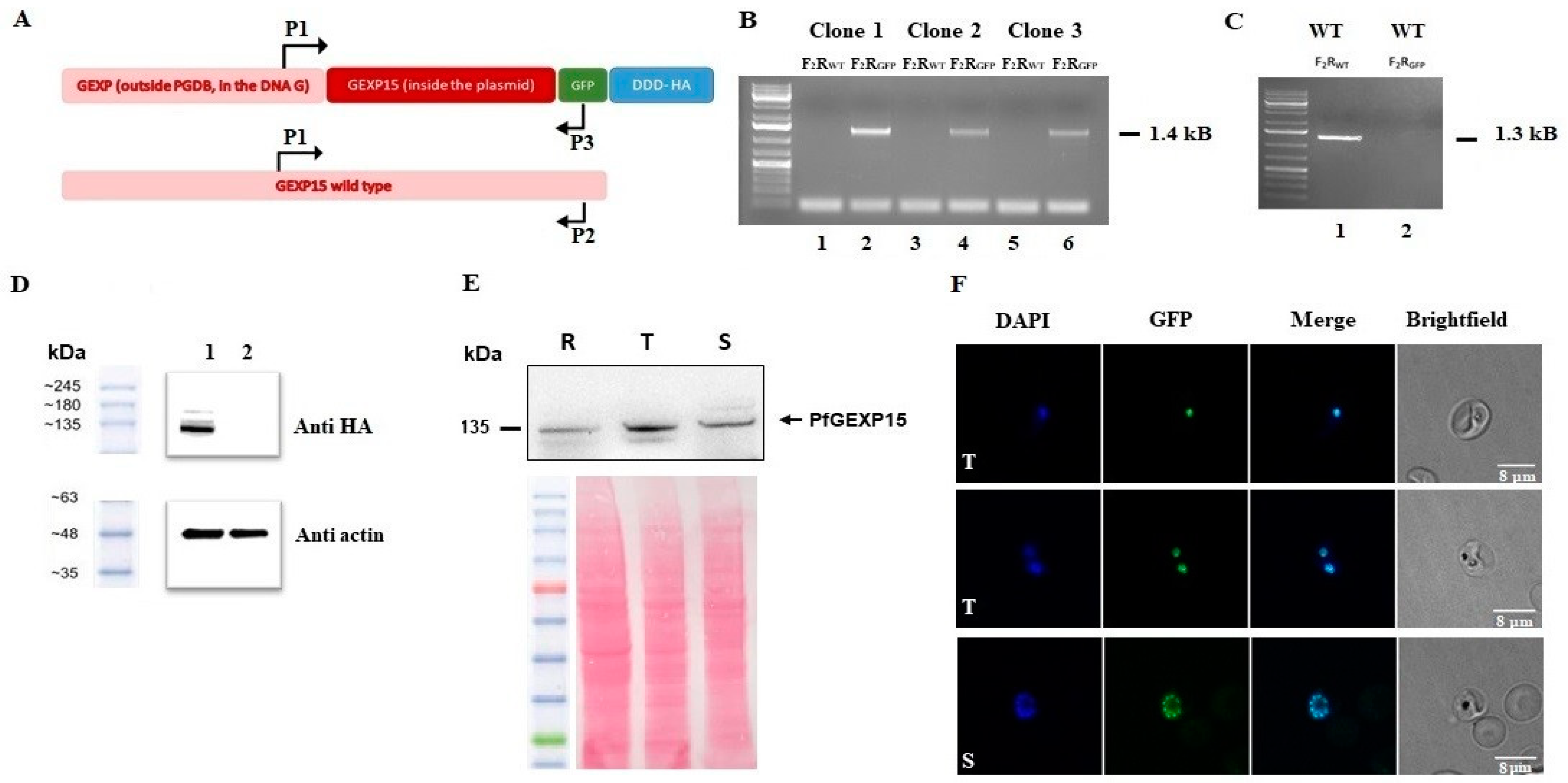
To further investigate the functional role of GEXP15, we attempted to conditionally knock down PfGEXP15 using a degradation domain since the protein was previously suggested as essential for the development of blood-stage parasites [
18]. Despite the integration of the degradation domain, confirmed by genotyping and immunoblotting, phenotypic analysis was not possible as the protein remained stable, suggesting that GEXP15 may be part of a large and stable complex. A previous study proposed that proteins not accessible to the proteasome for degradation could be a challenge for knockdown experiments [
34]. Other systems can be considered, such as the Cre-LoxP system, which can be used to excise the gene of interest [
35] or the
TetR-DOZI–aptamer module repressing translation [
36].
Next, we took advantage of the GFP and HA tagging of GEXP15 to follow up its localization throughout its intraerythrocytic stages. Confocal microscopy revealed that GEXP15 is highly expressed in late asexual stages, in agreement with previous transcriptomics data [
12], and is localized in the parasite nucleus. In contrast to the localization of PbGEXP15 in both the nucleus and cytoplasm [
7], this finding is similar to the human CD2BP2 localization [
37]. This potential difference between the two
Plasmodium species requires further investigation using electron microscopy or subcellular fractionation in order to confirm that the localization of GEXP15 is species-specific.
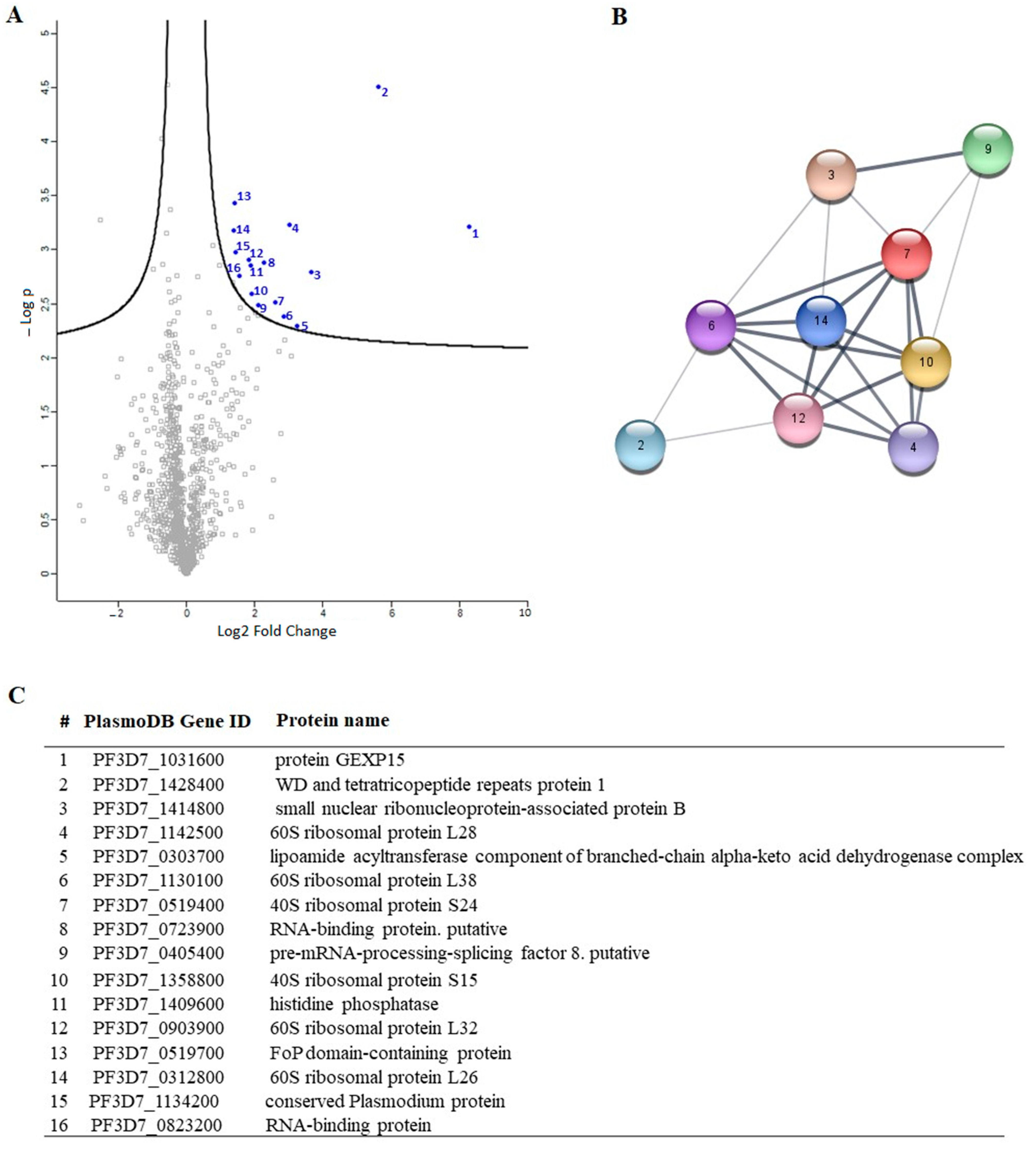
To gain a deeper understanding of the function of PfGEXP15, we profiled the GEXP15 interactome. A first approach based on immunoprecipitation experiments of endogenous tagged PfGEXP15-DDD-GFP-HA present in protein extracts by MS was applied to identify binding partners. This allowed the identification of 10 proteins related to one main functional group corresponding to the ribosomal complex and RNA-binding proteins.
Although PfPP1 was not detected in the PfGEXP15 IP/MS, the likelihood of this interaction via the RVxF motif was demonstrated by the use of complementary approaches such as Y2H, GST pull-down, and pull-down experiments (this study), confirming previous findings [
8,
9]. Supporting this is the fact that in
P. berghei parasites, PbGEXP15 was also detected among the top PP1-interacting proteins in both schizont and gametocyte stages [
9]. Further, the reciprocal IP/MS identified PbPP1 after PbGEXP15 immunoprecipitation [
7]. Using this approach, the lack of PfPP1 can be due to the fact that the complex PfGEXP15-PP1 is unstable at the time point examined and/or its association is transient at this stage.
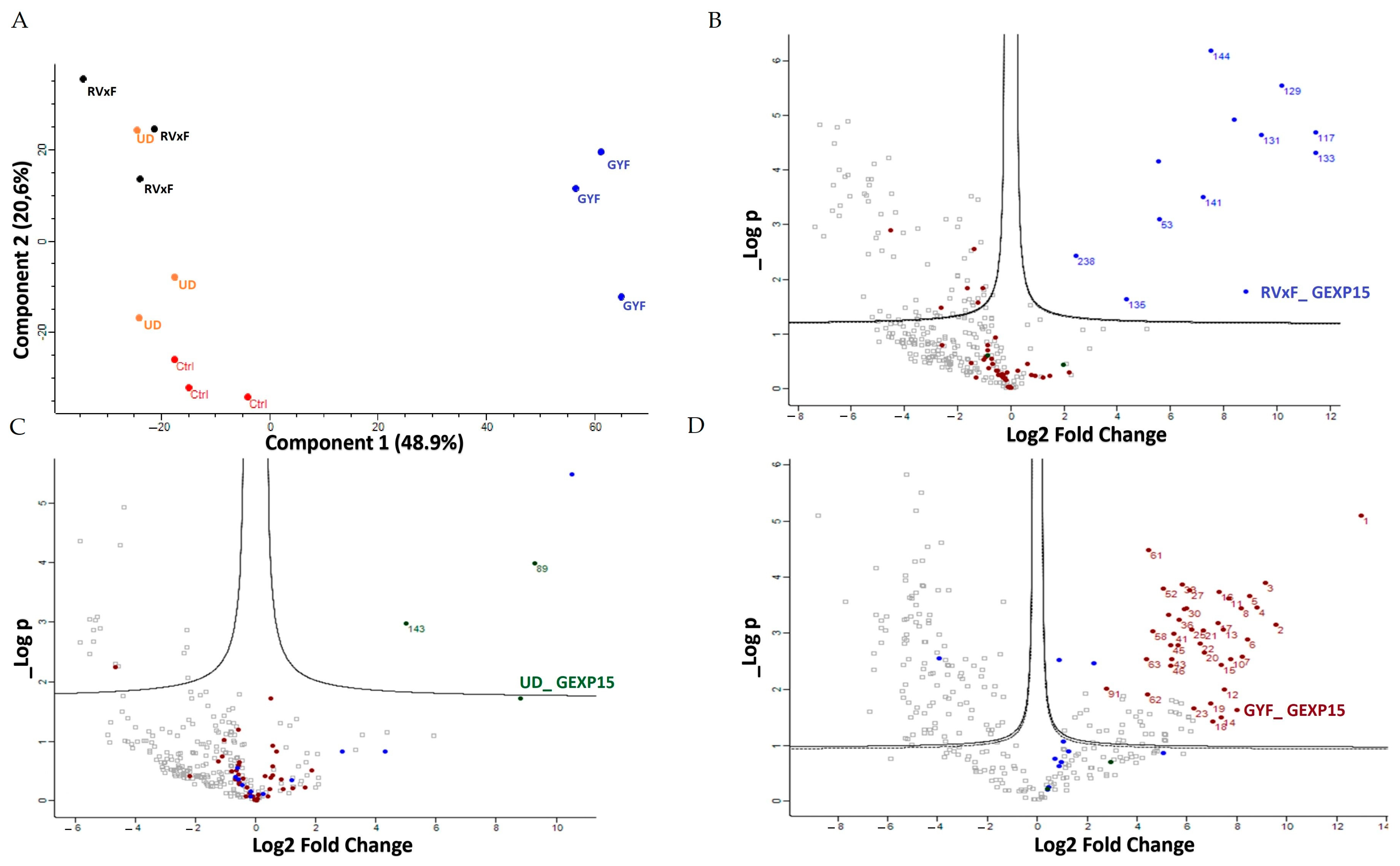
A second approach, using pull-down experiments with recombinant GYF-domain bound to beads and soluble protein extracts, revealed 29 proteins that belong to ribosomal subunits and ribosomal-associated proteins and of which three are shared with the ribosomal proteins detected in the IP-MS experiments. The limited subset of partners identified by IP and not by pull-down is not surprising as they represent different methods for interactome studies. It is known that the quantity of immunoprecipitated tagged protein, expected to be different from the quantity engaged in pull-down experiments, greatly affects MS identification. Hence, the results obtained herein can be complementary and, taken together, strongly suggest that PfGEXP15 is a ribosome-associated protein. More important is the fact that our data clearly revealed that the GYF-domain-containing protein of PfGEXP15 binds to ribosomal complex proteins, unlike the GYF domain of human CD2BP2 that has been shown to bind to spliceosomal proteins [
14]. This unexpected observation suggests that the GYF-containing proteins might have diverse interactomes according to their subcellular localization, the presence and availability of species-specific partners, and/or the subtle differences in amino acids within or around the GYF domain per se. This is supported by the fact that the binding partners of GEXP15 of Pb obtained by IP experiments are different from those of Pf as they belong to spliceosomal and proteasomal core complexes which could be, at least in part, attributed to the different localization of GEXP15 in both parasites.
A closer examination of the identified proteins in the PfGEXP15 interactome showed the presence of the ribosomal RNA processing 1 homolog b (PF3D7_1414800). Interestingly, an earlier study using quantitative affinity purification followed by mass spectrometry demonstrated that human RRP1B was the most abundant partner of PP1 [
38]. Moreover, it has been reported that nucleolar complexes contain both RRP1B and PP1 as components of pre-ribosomal subunit processing complexes [
39]. The potential involvement of PfGEXP15 in this RRP1B-PP1 complex could therefore be envisaged. Altogether, these findings are consistent with the fact that reversible phosphorylation events via PfPP1 likely contribute to fine-tuning ribosomal biogenesis.
Despite the fact that we have not obtained direct evidence on the impact of PfGEXP15 on intraerythrocytic parasite development as the knockdown approach based on protein degradation failed, our data showing the capacity of PfGEXP15 (1) to bind and regulate PfPP1c activity, essential for Plasmodium survival, through its N-terminal side and (2) to interact with the ribosomal protein complex via its C-terminal side, crucial for protein translation, strongly support the essentiality of PfGEXP15. Given the functional difference between human CD2BP2 and PfGEXP15, and particularly the specific partners of the latter, identified through the GYF domain-containing protein, it would be important to determine how they interact in order to exploit specific parasite PfGEXP15–ribosome interaction for malaria drug development.
In this context, we have already shown that peptides interrupting the interaction of PP1 to its regulators via the RVxF-binding motif were able to inhibit Pf growth in vitro [
40]. This proof of concept and validation of the binding of PfGEXP15 with PP1 and the ribosomal complex will open new opportunities to identify small inhibitors to disrupt this interaction network and the development of Pf.
4. Materials and Methods
4.1. Plasmid
Plasmid pGDB was a kind gift from Vasant Muralidharan. The integration plasmid, pGEXP15GDB, was synthetized by introducing a 984 bp fragment from the 3′ end of the GEXP15 ORF into pGDB between the XhoI/AvrII (New England Biolabs, Ipswich, MA, USA). PetDuet-1 was purchased from Novagen (Darmstadt, Germany).
4.2. Parasite Culture
The Pf3D7 strain was grown according to Trager and Jensen in RPMI 1640 medium with 10% human AB
+ serum, in the presence of O
+ erythrocytes [
41]. Cultures were maintained at 37 °C in a humidified atmosphere (5% CO
2, 5% O
2, and 90% N
2). Parasites were synchronized by successive rounds of 5% sorbitol treatment as described previously [
42]. In order to isolate total proteins, parasites from infected red blood cells were purified as previously described [
43].
4.3. MEME and FIMO Analysis
MEME Suite v5.5.1 (
https://meme-suite.org/meme/tools/meme accessed on 16 March 2023) was used on the full-length sequences of GEXP15 and CD2BP2 proteins to identify conserved motifs. A maximum of 5 motifs were searched for Pf, Pb, Tg, Sc, and human sequences with maximum widths of 30 and default parameters. For the 84 CD2BP2 proteins identified, a maximum of 7 motifs were searched with the same settings. FIMO v5.5.1 (
https://meme-suite.org/meme/doc/fimo.html accessed on 5 April 2023) was used to scan the RVxF motif among the 84 sequences using the consensus sequence [RK][RK][VI]X[FW] and default parameters.
4.4. 3D Modeling
The modeling of the PF3D7_1031600, NP_006101, and PBANKA_0515400 were carried out using Alphafold (
https://alphafold.ebi.ac.uk/ accessed on 24 November 2022). I-TASSER (
https://zhanggroup.org/I-TASSER/ accessed on 21 November 2022) was used additionally for the modelling of the two domains: UD (145 a.a.) and GYF (100 a.a.).
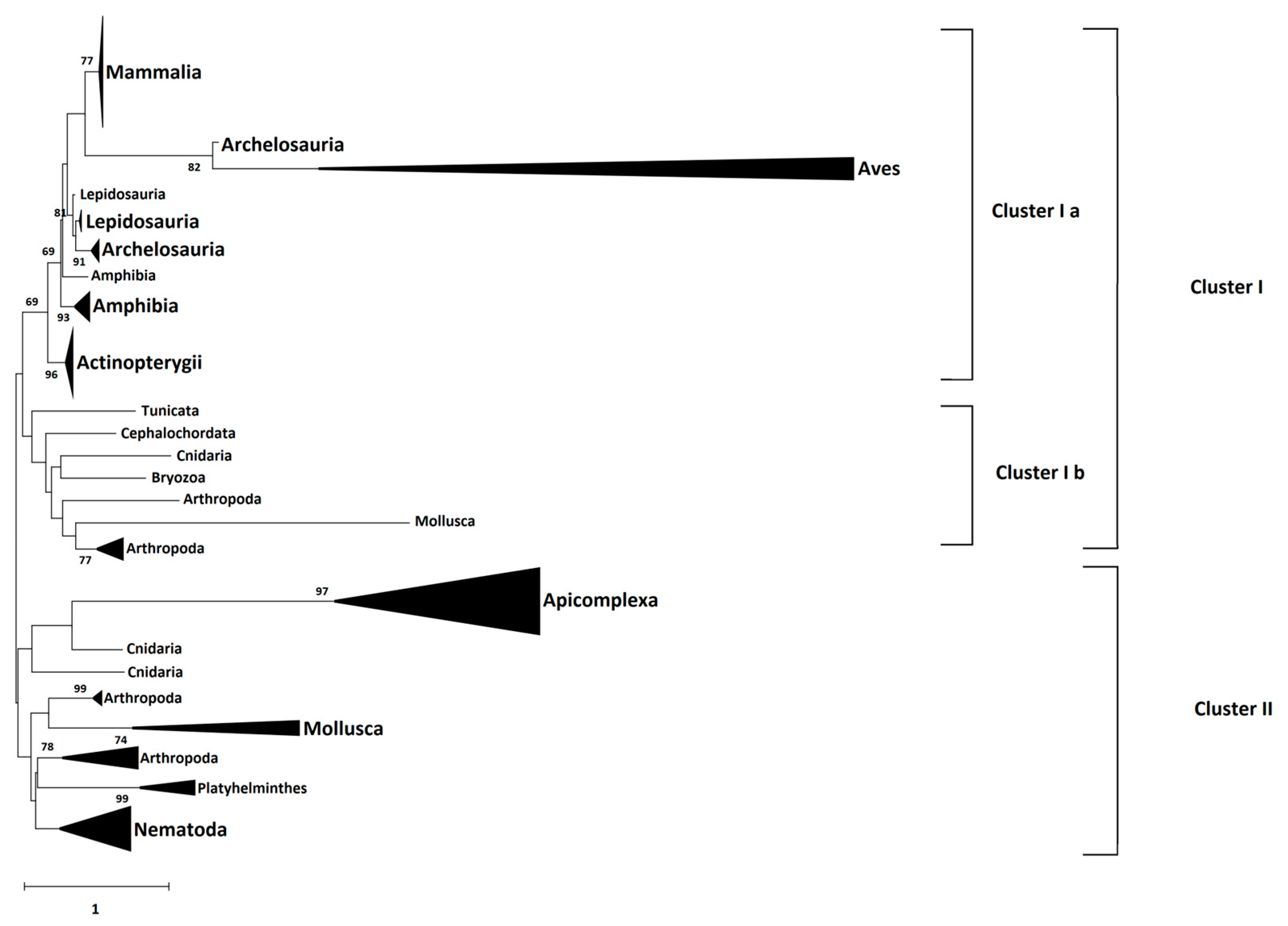
4.5. Phylogeny Analysis
The amino acid sequences of 66 identified CD2BP2 proteins were downloaded from the NCBI database (
https://www.ncbi.nlm.nih.gov/ accessed on 12 September 2022) as well as three Plasmodium GEXP15 sequences and six homologs from Apicomplexa parasites. The species and accession numbers of each sequence is provided in
Supplementary data sheet S2. Multiple sequence alignment of these full-length sequences was performed by Clustal Omega (
https://www.ebi.ac.uk/Tools/msa/clustalo/ accessed on 11 October 2022). Then, the Neighbor-Joining method and JTT matrix-based model, implemented in MEGA X software (Version 10.2.6), were used to build a phylogenetic tree from the sequence alignment. A gamma distribution equal to one with partial deletion was used. Reliability of internal branches was assessed using the bootstrapping method (500 bootstrap replicates).
4.6. Yeast Two-Hybrid Assays
pGADT7-PfGEXP15 RVxF was isolated from our initial yeast-two hybrid screening [
8]. Gal4-DBD-Laminin, Gal4-DBD-PfPP1c, and PfPP1c F255A/F256A were previously cloned in pGBKT7 [
40]. Y2H Gold (pGADT7-PfGEXP15 RVxF) and Y187 (pGBKT7 constructs) yeast strains (Clontech, California, USA) were mated on SD-LW media. Diploids were then selected on plates lacking leucine, tryptophan and histidine (SD-LWH), and adenine (SD-LWHA) after dilutions at 1:1, 1:25, and 1:50. Plates were incubated for 4–6 days at 30 °C.
4.7. GST Pull-Down Assays
The coding region of the three recombinant protein fragments were PCR amplified using genomic DNA with the following primers: (1) P4–P5 for the N-terminal fragment (21–546 bp); (2) P6–P7 for the central region (625–1242 bp); and (3) P8–P9 for the C terminal portion (1878–2445 bp) (
Supplementary Table S4). They were cloned into pETDuet-1 (Novagen, Darmstadt, Germany) using the In-Fusion HD Cloning system (Clontech, Mountain View, CA, USA) and transformed into One Shot
® BL21 Star™ (DE3) Chemically Competent
E. coli cells (Life Technologies, Carlsbad, CA, USA) (
Supplementary Table S5). The recombinant proteins were expressed in the presence of 0.5 mM IPTG at 16 °C overnight. Cells were harvested in non-denaturing buffer (20 mM Tris, 500 mM NaCl, 20 mM Imidazole, and protease inhibitor cocktail (Roche, Basel, Switzerland), pH 7.5) prior to sonication and ultracentrifugation. Then, the different pellets were resuspended for 30 min in denaturing buffer (20 mM Tris, 500 mM NaCl, 6 M Guanidine, 20 mM Imidazole, and protease inhibitor cocktail (Roche, Basel, Switzerland), pH 7.5). Recombinant proteins were purified by Ni
2+-NTA agarose beads (Macherey Nagel, Düren, Germany) and washed with 20 mM Tris, 500 mM NaCl, and 20 mM Imidazole, pH 7.5. His-tagged proteins were eluted from beads with buffer containing 20 mM Tris, 500 mM NaCl, and 600 mM Imidazole, pH 7.5, and then the imidazole was eliminated by dialysis. The purified recombinant proteins were analyzed by western blot with anti-His antibody (1:2000 dilution) (Qiagen, Hilden, Germany) followed by HRP-labeled anti-mouse IgG (1:50,000 dilution) and quantified with a Pierce™ BCA Protein Assay Kit (Life Technologies, Carlsbad, CA, USA). GST-PfPP1c and PfPP1c were produced as previously described [
7].
GST or GST-PfPP1c coupled with Glutathione-Sepharose beads (Sigma-Aldrich, Darmstadt, Germany) were saturated with 25 μg of BSA and incubated overnight at 4 °C with 2 μg of PfGEXP15 RVxF, UD, and GYF in binding buffer (20 mM Tris, 150 mM NaCl, 0.2 mM EDTA, 20 mM HEPES, 1 mM MnCl2, 1 mM DTT, 0.1% Triton X-100, 10% glycerol, protease inhibitor cocktail (Roche, Basel, Switzerland), and pH 7.5). After washes, proteins were analyzed by western blot, as well as 500 ng of PfGEXP15 RVxF, UD, and GYF used as inputs.
4.8. pNPP Phosphatase Assays
Different amounts of PfGEXP15 RVxF-, UD-, and GYF-containing proteins, described above, were preincubated with 40 pmol of PfPP1c for 30 min at 37 °C. Addition of p-nitrophenyl phosphate (pNPP) substrate (Sigma-Aldrich, Darmstadt, Germany) initiated the enzymatic reaction and after 1h of incubation, absorbance was measured at 405 nm (Thermo Scientific Multiskan FC, Marsiling Industrial Estate, Singapore). No phosphatase activity was detected with the different PfGEXP15 proteins in the absence of PP1. Two independent experiments were carried out in duplicate.
4.9. Transfection
To generate the PfGEXP15-HA-GFP parasite line, uninfected RBCs were transfected with 100 μg pGEXP15GDB vector then fed to wild type parasites. Drug pressure was applied 48 h after transfection, selecting for integration using 5 μM TMP (Sigma, Darmstadt, Germany) and 2.5 μg/mL Blasticidin (Calbiochem). Integration was detected after two rounds of BSD cycling after transfection. TMP was always present in the medium. Integrant clones were isolated by limiting dilution.
4.10. Genotype and Phenotype Analysis of Pf Transfectants
To confirm that transfected parasites contained the right integration, genomic DNA extracted (KAPA Express Extract, Kapa BioSystems, Dunedin, New Zealand) from wild or transfected parasites were analyzed by PCR using standard procedures with the primers P1–P3. Expression of the iKd PfGEXP15 protein was checked by western blotting using anti-HA (1/1000, Cell signaling C29F4, Massachusetts, USA) followed by anti-Rabbit IgG (1/20,000, Sigma, Darmstadt, Germany). Live parasites expressing PfGEXP15-GFP were analyzed by fluorescence microscopy as described below. To address the phenotype of transgenic parasites, cultures highly enriched with late trophozoites (>80%) were washed 6 times then set up ± TMP at 1% of infected red blood cells. The parasitemia were monitored up for 12 days (covering 6 life cycle) on a daily basis. After 3 and 5 cycles, viable parasites were checked for PfGEXP15 expression by live microscopy and immunoblot assays.
4.11. Immunoblot Assays
Parasites were suspended in Laemmli buffer and total proteins were subjected to electrophoresis in a 10% polyacrylamide gel. The proteins were transferred onto a nitrocellulose membrane (Amersham Protran 0.45 μm NC). The membrane was blocked with 5% milk (non-fat milk powder dissolved in PBS) and probed with primary antibodies (rabbit anti-HA, 1/1000 or mouse anti-His, 1/2000) diluted in the blocking buffer. The primary antibodies were followed by respective species-specific secondary antibodies conjugated to HRP (anti-rabbit, 1/20,000, Sigma) or (anti-mouse, 1/20,000, Rockland). The antibody incubations were followed by thorough washing using PBS tween 0.4%. The membranes were visualized using Dura/ Femto western blotting substrate.
4.12. Fluorescence Microscopy
Transgenic and parental parasites were washed then fixed with 4% paraformaldehyde and 0.0075% glutaraldehyde for 15 min at 4 °C. After PBS washing, cells were settled on Poly-L-lysine coated coverslips. The coverslips were mounted in Mowiol with DAPI (1 μg/mL) and multipoint-confocal imaging was performed with a spinning disk Live SR (stand Nikon Ti-2 combined with Live-SR module Gataca Systems, Massy, France). Figures were produced using ImageJ/Fiji software (ImageJ 1.54f, National Institutes of Health, USA).
4.13. Pull-Down Assays
For pull-down experiments, the RVxF-containing protein, described above, was used. For the other two recombinant proteins, shorter fragments were synthesized in order to retain the minimal functional domains based on sequence and structure analyses (UD:853–1266 bp; GYF:2053–2347 bp) (
Supplementary Table S5).
The expression of His6-motifs was carried out in the E. coli BL21 strain in the presence of 0.5 mM IPTG at 37 °C for 2 h. Cells were harvested in lysis buffer (20 mM Tris, 150 mM NaCl, 20 mM Imidazole, Triton 1%, Lysozyme 1 mg/50 mL, DNase I, and protease inhibitor cocktail (Roche, Basel, Switzerland), pH 7.5). Recombinant proteins were purified according to manufacturer’s instructions by Ni2+-NTA agarose beads (QIAGEN, Hilden, Germany). Washing steps were performed with a buffer containing 20 mM Tris, 150 mM NaCl, and 20 mM imidazole, pH 7.5. Three additional washing steps with a buffer containing 20 mM Tris, 150 mM NaCl, 0.5% Triton X-100, and protease inhibitor cocktail (Roche, Basel, Switzerland), pH 7.5 were done before adding the soluble proteins to parasite extracts.
For the pull-down experiment, trophozoites/schizonts of parental wild-type parasites were suspended in 50 mM Tris, 0.5% Triton X-100, 150 mm NaCl, and protease inhibitor cocktail (Roche), pH 7.5. After ten consecutive freezing–thawing cycles and sonication, soluble fractions were obtained after repeated centrifugations at 13,000 rpm at 4 °C.
The agarose nickel beads coated with the recombinant proteins were mixed overnight at 4 °C with parasite soluble extracts in 20 mM Tris, 150 mM NaCl, 0.5% Triton X-100, and protease inhibitor cocktail (Roche, Basel, Switzerland), pH 7.5.
The beads were washed and elution was performed in Laemmli buffer. Then, after 3 min at 95 °C, samples were loaded on a 4–20% SDS-PAGE for western blot or mass spectrometry analyses. Western blots were carried out probed with anti-His mAb (1:1000, Invitrogen, Waltham, MA, USA) followed by anti-mice IgG-HRP (1:20,000, Sigma-Aldrich).
4.14. Sample Preparation and Immunoprecipitation
Pf-enriched trophozoite and schizont cultures of PfGEXP15-GFP-DDD-HA or parental wildtype strain (control) were used for protein extracts as described above.
Immunoprecipitation experiments were performed using 3 biological replicates of each strain. Each biological replicate contained 10 isolated pellets of trophozoites and schizonts, each purified from one culture flask of 75 cm
2. Soluble protein extractions and immunoprecipitation assays were performed as previously described [
7]. Purified parasites of each strain were suspended in 50 mM Tris, 0.5% Triton X-100, 150 mm NaCl, and protease inhibitor cocktail (Roche, Basel, Switzerland), pH 7.5. After ten consecutive freezing–thawing cycles and sonication, soluble fractions were obtained after repeated centrifugations at 13,000 rpm at 4 °C. These soluble fractions were incubated with GFP-Trap magnetic agarose (ChromoTek, Martinsried, Germany) overnight at 4 °C on a rotating wheel. The beads were washed 10 times with washing buffer containing 20 mM Tris,150 mM NaCl, 0.5% Triton X-100, and protease inhibitor cocktail (Roche, Basel, Switzerland) at pH 7.5. Elution was performed in Laemmli buffer.
4.15. Sample Preparation for Mass Spectrometry
S-TrapTM micro spin column (Protifi, Huntington, WV, USA) digestion was performed on immunoprecipitation eluates and pull-down eluates according to the manufacturer’s instructions. Briefly, samples were supplemented with 20% SDS to a final concentration of 5%, reduced with 20 mM TCEP (Tris(2-carboxyethyl) phosphine hydrochloride), and alkylated with 50 mM CAA (chloroacetamide) for 5 min at 95 °C. Aqueous phosphoric acid was then added to a final concentration of 2.5% followed by the addition of S-Trap binding buffer (90% aqueous methanol, 100mM TEAB, pH 7.1). The mixtures were then loaded on S-Trap columns. Five washes were performed for thorough SDS elimination. Samples were digested with 2 µg of trypsin (Promega, Madison, WI, USA) at 47 °C for 2 h. After elution, peptides were vacuum dried and resuspended in 2% ACN, 0.1% formic acid in HPLC-grade water prior to MS analysis.
4.16. NanoLC-MS/MS Protein Identification and Quantification
The tryptic peptides were resuspended in 30 µL and an amount of 400 ng was injected on a nanoElute (Bruker Daltonics, Bremen, Germany) HPLC (high-performance liquid chromatography) system coupled to a timsTOF Pro (Bruker Daltonics, Bremen, Germany) mass spectrometer. HPLC separation (Solvent A: 0.1% formic acid in water; Solvent B: 0.1% formic acid in acetonitrile) was carried out at 250 nL/min using a packed emitter column (C18, 25 cm × 75 μm 1.6 μm) (Ion Optics, Melbourne, Australia) using a 40 min gradient elution (2 to 11% solvent B during 19 min; 11 to 16% during 7 min; 16% to 25% during 4 min; 25% to 80% for 3 min; and, finally, 80% for 7 min to wash the column). Mass spectrometric data were acquired using the parallel accumulation serial fragmentation (PASEF) acquisition method in DDA (data-dependent analysis) mode. The measurements were carried out over the m/z range from 100 to 1700 Th. The range of ion mobilities values were from 0.7 to 1.1 V s/cm2 (1/k0). The total cycle time was set to 1.2 s and the number of PASEF MS/MS scans was set to 6.
Data analysis was performed using MaxQuant software version 2.1.3.0 and searched with the Andromeda search engine against the TrEMBL/Swiss-Prot Pf 3D7 database downloaded from Uniprot on 10 October 2022 (5392 entries) and the
E. coli BL21-DE3 database downloaded from Uniprot on 10 October 2022 (4173 entries). To search parent mass and fragment ions, we set a mass deviation of 10 ppm for the main search and 40 ppm, respectively. The minimum peptide length was set to 7 amino acids and strict specificity for trypsin cleavage was required, allowing up to 2 missed cleavage sites. Carbamidomethylation (Cys) was set as fixed modification, whereas oxidation (Met) and N-term acetylation (Prot N-term) were set as variable modifications. The false discovery rates (FDRs) at the peptide and protein levels were set to 1%. Scores were calculated in MaxQuant as described previously [
44]. The reverse and common contaminants hits were removed from MaxQuant output as well as the protein only identified by site. Proteins were quantified according to the MaxQuant label-free algorithm using LFQ intensities, and protein quantification was obtained using at least 1 peptide per protein. Matching between runs was allowed only with IP samples.
Statistical and bioinformatic analysis, including heatmaps, profile plots, and clustering, were performed with Perseus software (version 1.6.15.0) freely available at
www.perseus-framework.org accessed on 10 October 2022 [
45]. For statistical comparison, we set four groups, each containing up to 3 biological replicates for the pull-down samples (Control, RVxF, UD, GYF). For the IP samples, we set two groups with 3 biological replicates each (Control, GEXP15). We then filtered the data to keep only proteins with at least 3 and 2 valid values in at least one group for pull-down and IP experiments, respectively. Next, the data were imputed to fill missing data points by creating a Gaussian distribution of random numbers with a standard deviation of 33% relative to the standard deviation of the measured values and using 3 and 1.8 SD downshift of the mean to simulate the distribution of low signal values for pull-down and IP datasets, respectively. We then performed an ANOVA test (FDR < 0.05, S0 = 1) for the pull-down samples and a statistical
t-test (FDR < 0.05, S0 = 0.1) for IP samples. Hierarchical clustering of proteins that survived the test was performed in Perseus on LFQ intensities expressed on a logarithmic scale after z-score normalization of the data using Euclidean distances.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







