

Insights into the Inhibitory Mechanisms of the Covalent Drugs for DNMT3A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
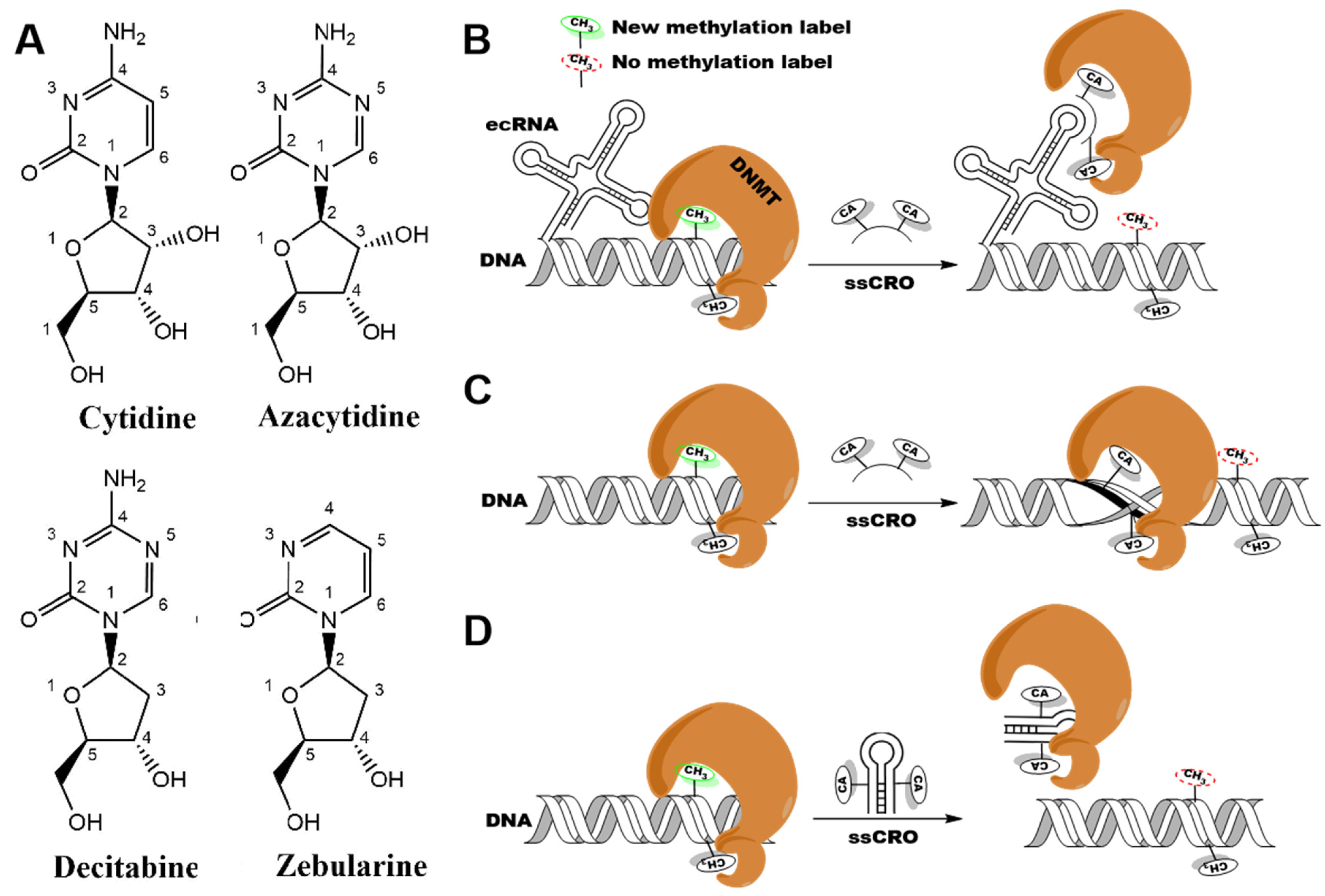
:1. Introduction
2. Result
2.1. The Characterization of the Pre-Reaction State
2.2. Deprotonation of the Cys
2.3. The S-C Attack
2.4. Methyl Transfer Process
2.5. The MD of the IM3 States
3. Discussion
4. Materials and Methods
4.1. MD Input Preparation
4.2. MD Process
4.3. QM/MM Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riddihough, G.; Zahn, L.M. What is epigenetics? Science 2010, 330, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Wang, R.Y. 5-Methylcytosine in eukaryotic DNA. Science 1981, 212, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Nelkin, B.D.; Celano, P.; Yen, R.W.; Falco, J.P.; Hamilton, S.R.; Baylin, S.B. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc. Natl. Acad. Sci. USA 1991, 88, 3470–3474. [Google Scholar] [CrossRef]
- Patra, S.K.; Patra, A.; Zhao, H.; Dahiya, R. DNA methyltransferase and demethylase in human prostate cancer. Mol. Carcinog. 2002, 33, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.l.; Tubiana-Hulin, M.; Lidereau, R.; Bièche, I. Expression analysis of estrogen receptor α coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 2003, 9, 1259–1266. [Google Scholar]
- Oh, B.K.; Kim, H.; Park, H.J.; Shim, Y.H.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 2007, 20, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Melki, J.R.; Warnecke, P.; Vincent, P.C.; Clark, S.J. Increased DNA methyltransferase expression in leukaemia. Leukemia 1998, 12, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Nikula, K.J.; Baylin, S.B.; Issa, J.P. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 4045–4050. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Zeng, Y.; Ren, R.; Kaur, G.; Hardikar, S.; Ying, Z.; Babcock, L.; Gupta, E.; Zhang, X.; Chen, T.; Cheng, X. The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b-mediated DNA methylation. Genes. Dev. 2020, 34, 1546–1558. [Google Scholar] [CrossRef]
- Xu, P.; Hu, G.; Luo, C.; Liang, Z. DNA methyltransferase inhibitors: An updated patent review (2012–2015). Expert. Opin. Ther. Pat. 2016, 26, 1017–1030. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Agrawal, K.; Das, V.; Vyas, P.; Hajduch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA methylation with small molecules: What’s next? Miniperspective. J. Med. Chem. 2015, 58, 2569–2583. [Google Scholar] [CrossRef]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Marquez, V.E.; Mao, D.T.; Haines, D.R.; Mccormack, J.J. Synthesis of Pyrimidin-2-One Nucleosides as Acid-Stable Inhibitors of Cytidine Deaminase. J. Med. Chem. 1986, 29, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Chauhan, M.S.; Manik, R.S.; Palta, P.; Singla, S.K. Comparison the effects of 5-Aza-2’-deoxycytidine and zebularine on the in vitro development, blastocyst quality, methylation pattern and conception rate on handmade cloned buffalo embryos. Reprod. Domest. Anim. 2023, 58, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Chuang, J.C.; Byun, H.M.; Egger, G.; Yang, A.S.; Dubeau, L.; Long, T.; Laird, P.W.; Marquez, V.E.; Jones, P.A. Long-term Epigenetic Therapy with Oral Zebularine Has Minimal Side Effects and Prevents Intestinal Tumors in Mice. Cancer Prev. Res. 2008, 1, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gabbara, S.; Bhagwat, A.S. The Mechanism of Inhibition of DNA (Cytosine-5-)-Methyltransferases by 5-Azacytosine Is Likely to Involve Methyl Transfer to the Inhibitor. Biochem. J. 1995, 307 Pt 1, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowler, E.H.; Bell, J.; Divecha, N.; Skipp, P.; Ewing, R.M. Proteomic Analysis of Azacitidine-Induced Degradation Profiles Identifies Multiple Chromatin and Epigenetic Regulators Including Uhrf1 and Dnmt1 as Sensitive to Azacitidine. J. Proteome Res. 2019, 18, 1032–1042. [Google Scholar] [CrossRef]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.M.; Ausseil, F.; Vispe, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef]
- Cowan, L.A.; Talwar, S.; Yang, A.S. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics 2010, 2, 71–86. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Medina-Franco, J.L.; Mendez-Lucio, O.; Duenas-Gonzalez, A.; Yoo, J. Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today 2015, 20, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.; Jeltsch, A.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: A chemical and therapeutic patent overview and selected clinical studies. Expert. Opin. Ther. Pat. 2012, 22, 1427–1442. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.J.; Wang, Y.L.; Zhou, W.; Li, S.S.; Peng, J.L.; Shi, Z.; Hu, J.C.; Liu, Y.C.; Ding, H.; Lin, Y.Y.; et al. Identifying Novel Selective Non-Nucleoside DNA Methyltransferase 1 Inhibitors through Docking-Based Virtual Screening. J. Med. Chem. 2014, 57, 9028–9041. [Google Scholar] [CrossRef]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.Z.; Wang, Y.M.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Fagan, R.L.; Cryderman, D.E.; Kopelovich, L.; Wallrath, L.L.; Brenner, C. Laccaic Acid A Is a Direct, DNA-competitive Inhibitor of DNA Methyltransferase 1. J. Biol. Chem. 2013, 288, 23858–23867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, J.; Zinovjev, K.; Swiderek, K.; Roca, M.; Tunon, I. Unraveling the Reaction Mechanism of Enzymatic C5-Cytosine Methylation of DNA. A Combined Molecular Dynamics and QM/MM Study of Wild Type and Gln119 Variant. ACS Catal. 2016, 6, 3262–3276. [Google Scholar] [CrossRef] [Green Version]
- Aranda, J.; Attana, F.; Tunon, I. Molecular Mechanism of Inhibition of DNA Methylation by Zebularine. ACS Catal. 2017, 7, 1728–1732. [Google Scholar] [CrossRef]
- Liu, L.; Shi, T.; Houk, K.N.; Zhao, Y.L. Understanding the R882H mutation effects of DNA methyltransferase DNMT3A: A combination of molecular dynamics simulations and QM/MM calculations. RSC Adv. 2019, 9, 31425–31434. [Google Scholar] [CrossRef]
- Yang, W.; Zhuang, J.; Li, C.; Cheng, G.-J. Unveiling the Methyl Transfer Mechanisms in the Epigenetic Machinery DNMT3A-3L: A Comprehensive Study Integrating Assembly Dynamics with Catalytic Reactions. Comput. Struct. Biotechnol. J. 2023, 21, 2086–2099. [Google Scholar] [CrossRef]
- Lamiable-Oulaidi, F.; Harijan, R.K.; Shaffer, K.J.; Crump, D.R.; Sun, Y.; Du, Q.; Gulab, S.A.; Khan, A.A.; Luxenburger, A.; Woolhouse, A.D.; et al. Synthesis and Characterization of Transition-State Analogue Inhibitors against Human DNA Methyltransferase 1. J. Med. Chem. 2022, 65, 5462–5494. [Google Scholar] [CrossRef]
- Gao, Q.; Steine, E.J.; Barrasa, M.I.; Hockemeyer, D.; Pawlak, M.; Fu, D.; Reddy, S.; Bell, G.W.; Jaenisch, R. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc. Natl. Acad. Sci. USA 2011, 108, 18061–18066. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef]
- Zhang, X.; Bruice, T.C. The mechanism of M.HhaI DNA C5 cytosine methyltransferase enzyme: A quantum mechanics/molecular mechanics approach. Proc. Natl. Acad. Sci. USA 2006, 103, 6148–6153. [Google Scholar] [CrossRef] [PubMed]
- Zangi, R.; Arrieta, A.; Cossio, F.P. Mechanism of DNA methylation: The double role of DNA as a substrate and as a cofactor. J. Mol. Biol. 2010, 400, 632–644. [Google Scholar] [CrossRef]
- Yang, J.; Lior-Hoffmann, L.; Wang, S.; Zhang, Y.; Broyde, S. DNA cytosine methylation: Structural and thermodynamic characterization of the epigenetic marking mechanism. Biochemistry 2013, 52, 2828–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef]
- Shieh, F.-K.; Reich, N.O. AdoMet-dependent methyl-transfer: Glu119 is essential for DNA C5-cytosine methyltransferase M. HhaI. J. Mol. Biol. 2007, 373, 1157–1168. [Google Scholar] [CrossRef]
- Sarris, J.; Price, L.H.; Carpenter, L.L.; Tyrka, A.R.; Ng, C.H.; Papakostas, G.I.; Jaeger, A.; Fava, M.; Mischoulon, D. Is S-adenosyl methionine (SAMe) for depression only effective in males? A re-analysis of data from a randomized clinical trial. Pharmacopsychiatry 2015, 48, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D. S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: A clinician-oriented review of research. J. Clin. Psychiatry 2017, 78, 18881. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.F.; Yang, W.; Shi, Y.H.; Cheng, X.R.; Le, G.W. Structure-based approach for the study of thyroid hormone receptor binding affinity and subtype selectivity. J. Biomol. Struct. Dyn. 2016, 34, 2251–2267. [Google Scholar] [CrossRef]
- Wang, F.; Yang, W.; Hu, X. Discovery of high affinity receptors for dityrosine through inverse virtual screening and docking and molecular dynamics. Int. J. Mol. Sci. 2018, 20, 115. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.F.; Yang, W.; Li, R.; Sui, Z.H.; Cheng, G.J.; Zhou, B. Molecular description of pyrimidine-based inhibitors with activity against FAK combining 3D-QSAR analysis, molecular docking and molecular dynamics. Arab. J. Chem. 2021, 14, 103144. [Google Scholar] [CrossRef]
- Wang, F.; Yang, W.; Liu, H.; Zhou, B. Identification of the structural features of quinazoline derivatives as EGFR inhibitors using 3D-QSAR modeling, molecular docking, molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2022, 40, 11125–11140. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Riley, B.T.; Lei, X.; Porebski, B.T.; Kass, I.; Buckle, A.M.; McGowan, S. Mapping the Pathway and Dynamics of Bestatin Inhibition of the Plasmodium falciparum M1 Aminopeptidase PfA-M1. ChemMedChem 2018, 13, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Riley, B.T.; Lei, X.; Porebski, B.T.; Kass, I.; Buckle, A.M.; McGowan, S. Generation of AMBER force field parameters for zinc centres of M1 and M17 family aminopeptidases. J. Biomol. Struct. Dyn. 2018, 36, 2595–2604. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Case, D.A.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; Gohlke, H.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges—The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Ivani, I.; Dans, P.D.; Noy, A.; Perez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goni, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Smith, W.; Forester, T. ChemShell-amodular software package for QM/MM simulation. J. Mol. Graphics Modell. 1996, 14, 136–141. [Google Scholar] [CrossRef]
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to molecular simulation. Mol. Simulat 2002, 28, 385–471. [Google Scholar] [CrossRef]
- De Vries, A.H.; Sherwood, P.; Collins, S.J.; Rigby, A.M.; Rigutto, M.; Kramer, G.J. Zeolite structure and reactivity by combined quantum-chemical—Classical calculations. J. Phys. Chem. B 1999, 103, 6133–6141. [Google Scholar] [CrossRef]
- Billeter, S.R.; Turner, A.J.; Thiel, W. Linear scaling geometry optimisation and transition state search in hybrid delocalised internal coordinates. Phys. Chem. Chem. Phys. 2000, 2, 2177–2186. [Google Scholar] [CrossRef]
- Senn, H.M.; Kastner, J.; Breidung, J.; Thiel, W. Finite-temperature effects in enzymatic reactions—Insights from QM/MM free-energy simulations. Can. J. Chem. 2009, 87, 1322–1337. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, Y. Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: An ab initio QM/MM-FE study with multiple initial structures. J. Am. Chem. Soc. 2006, 128, 1272–1278. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, S.; Thiel, W. Enzymatic Hydroxylation in p-Hydroxybenzoate Hydroxylase: A Case Study for QM/MM Molecular Dynamics. J. Chem. Theory Comput. 2005, 1, 494–505. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, W.; Zhuang, J.; Li, C.; Bai, C.; Cheng, G. Insights into the Inhibitory Mechanisms of the Covalent Drugs for DNMT3A. Int. J. Mol. Sci. 2023, 24, 12652. https://doi.org/10.3390/ijms241612652
Yang W, Zhuang J, Li C, Bai C, Cheng G. Insights into the Inhibitory Mechanisms of the Covalent Drugs for DNMT3A. International Journal of Molecular Sciences. 2023; 24(16):12652. https://doi.org/10.3390/ijms241612652
Chicago/Turabian StyleYang, Wei, Jingyuan Zhuang, Chen Li, Chen Bai, and Guijuan Cheng. 2023. "Insights into the Inhibitory Mechanisms of the Covalent Drugs for DNMT3A" International Journal of Molecular Sciences 24, no. 16: 12652. https://doi.org/10.3390/ijms241612652